All results from a given calculation for C12H8 (biphenylene)
using model chemistry: QCISD(T)/cc-pVDZ
19 10 17 12 22
States and conformations
| State |
Conformation |
minimum conformation |
conformer description |
state description |
| 1 |
1 |
yes |
D2H |
1Ag |
Energy calculated at QCISD(T)/cc-pVDZ
| | hartrees |
|---|
| Energy at 0K | -460.726383 |
| Energy at 298.15K | |
| HF Energy | -459.040442 |
| Nuclear repulsion energy | 583.719831 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at QCISD(T)/cc-pVDZ
Geometric Data calculated at QCISD(T)/cc-pVDZ
Point Group is D2h
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.000 |
0.720 |
0.763 |
| C2 |
0.000 |
0.720 |
-0.763 |
| C3 |
0.000 |
-0.720 |
0.763 |
| C4 |
0.000 |
-0.720 |
-0.763 |
| C5 |
0.000 |
1.463 |
1.934 |
| C6 |
0.000 |
0.702 |
3.150 |
| C7 |
0.000 |
-0.702 |
3.150 |
| C8 |
0.000 |
-1.463 |
1.934 |
| C9 |
0.000 |
1.463 |
-1.934 |
| C10 |
0.000 |
0.702 |
-3.150 |
| C11 |
0.000 |
-0.702 |
-3.150 |
| C12 |
0.000 |
-1.463 |
-1.934 |
| H13 |
0.000 |
2.560 |
1.954 |
| H14 |
0.000 |
1.234 |
4.111 |
| H15 |
0.000 |
-1.234 |
4.111 |
| H16 |
0.000 |
-2.560 |
1.954 |
| H17 |
0.000 |
2.560 |
-1.954 |
| H18 |
0.000 |
1.234 |
-4.111 |
| H19 |
0.000 |
-1.234 |
-4.111 |
| H20 |
0.000 |
-2.560 |
-1.954 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
C3 |
C4 |
C5 |
C6 |
C7 |
C8 |
C9 |
C10 |
C11 |
C12 |
H13 |
H14 |
H15 |
H16 |
H17 |
H18 |
H19 |
H20 |
| C1 | | 1.5251 | 1.4398 | 2.0974 | 1.3875 | 2.3880 | 2.7795 | 2.4777 | 2.7973 | 3.9131 | 4.1635 | 3.4697 | 2.1925 | 3.3881 | 3.8770 | 3.4899 | 3.2814 | 4.9010 | 5.2509 | 4.2591 |
C2 | 1.5251 | | 2.0974 | 1.4398 | 2.7973 | 3.9131 | 4.1635 | 3.4697 | 1.3875 | 2.3880 | 2.7795 | 2.4777 | 3.2814 | 4.9010 | 5.2509 | 4.2591 | 2.1925 | 3.3881 | 3.8770 | 3.4899 | C3 | 1.4398 | 2.0974 | | 1.5251 | 2.4777 | 2.7795 | 2.3880 | 1.3875 | 3.4697 | 4.1635 | 3.9131 | 2.7973 | 3.4899 | 3.8770 | 3.3881 | 2.1925 | 4.2591 | 5.2509 | 4.9010 | 3.2814 | C4 | 2.0974 | 1.4398 | 1.5251 | | 3.4697 | 4.1635 | 3.9131 | 2.7973 | 2.4777 | 2.7795 | 2.3880 | 1.3875 | 4.2591 | 5.2509 | 4.9010 | 3.2814 | 3.4899 | 3.8770 | 3.3881 | 2.1925 | C5 | 1.3875 | 2.7973 | 2.4777 | 3.4697 | | 1.4347 | 2.4839 | 2.9266 | 3.8683 | 5.1413 | 5.5267 | 4.8507 | 1.0972 | 2.1893 | 3.4661 | 4.0237 | 4.0401 | 6.0499 | 6.6199 | 5.5954 | C6 | 2.3880 | 3.9131 | 2.7795 | 4.1635 | 1.4347 | | 1.4049 | 2.4839 | 5.1413 | 6.3010 | 6.4557 | 5.5267 | 2.2097 | 1.0980 | 2.1615 | 3.4752 | 5.4322 | 7.2813 | 7.5156 | 6.0583 | C7 | 2.7795 | 4.1635 | 2.3880 | 3.9131 | 2.4839 | 1.4049 | | 1.4347 | 5.5267 | 6.4557 | 6.3010 | 5.1413 | 3.4752 | 2.1615 | 1.0980 | 2.2097 | 6.0583 | 7.5156 | 7.2813 | 5.4322 | C8 | 2.4777 | 3.4697 | 1.3875 | 2.7973 | 2.9266 | 2.4839 | 1.4347 | | 4.8507 | 5.5267 | 5.1413 | 3.8683 | 4.0237 | 3.4661 | 2.1893 | 1.0972 | 5.5954 | 6.6199 | 6.0499 | 4.0401 | C9 | 2.7973 | 1.3875 | 3.4697 | 2.4777 | 3.8683 | 5.1413 | 5.5267 | 4.8507 | | 1.4347 | 2.4839 | 2.9266 | 4.0401 | 6.0499 | 6.6199 | 5.5954 | 1.0972 | 2.1893 | 3.4661 | 4.0237 | C10 | 3.9131 | 2.3880 | 4.1635 | 2.7795 | 5.1413 | 6.3010 | 6.4557 | 5.5267 | 1.4347 | | 1.4049 | 2.4839 | 5.4322 | 7.2813 | 7.5156 | 6.0583 | 2.2097 | 1.0980 | 2.1615 | 3.4752 | C11 | 4.1635 | 2.7795 | 3.9131 | 2.3880 | 5.5267 | 6.4557 | 6.3010 | 5.1413 | 2.4839 | 1.4049 | | 1.4347 | 6.0583 | 7.5156 | 7.2813 | 5.4322 | 3.4752 | 2.1615 | 1.0980 | 2.2097 | C12 | 3.4697 | 2.4777 | 2.7973 | 1.3875 | 4.8507 | 5.5267 | 5.1413 | 3.8683 | 2.9266 | 2.4839 | 1.4347 | | 5.5954 | 6.6199 | 6.0499 | 4.0401 | 4.0237 | 3.4661 | 2.1893 | 1.0972 | H13 | 2.1925 | 3.2814 | 3.4899 | 4.2591 | 1.0972 | 2.2097 | 3.4752 | 4.0237 | 4.0401 | 5.4322 | 6.0583 | 5.5954 | | 2.5326 | 4.3644 | 5.1206 | 3.9082 | 6.2089 | 7.1544 | 6.4417 | H14 | 3.3881 | 4.9010 | 3.8770 | 5.2509 | 2.1893 | 1.0980 | 2.1615 | 3.4661 | 6.0499 | 7.2813 | 7.5156 | 6.6199 | 2.5326 | | 2.4673 | 4.3644 | 6.2089 | 8.2229 | 8.5850 | 7.1544 | H15 | 3.8770 | 5.2509 | 3.3881 | 4.9010 | 3.4661 | 2.1615 | 1.0980 | 2.1893 | 6.6199 | 7.5156 | 7.2813 | 6.0499 | 4.3644 | 2.4673 | | 2.5326 | 7.1544 | 8.5850 | 8.2229 | 6.2089 | H16 | 3.4899 | 4.2591 | 2.1925 | 3.2814 | 4.0237 | 3.4752 | 2.2097 | 1.0972 | 5.5954 | 6.0583 | 5.4322 | 4.0401 | 5.1206 | 4.3644 | 2.5326 | | 6.4417 | 7.1544 | 6.2089 | 3.9082 | H17 | 3.2814 | 2.1925 | 4.2591 | 3.4899 | 4.0401 | 5.4322 | 6.0583 | 5.5954 | 1.0972 | 2.2097 | 3.4752 | 4.0237 | 3.9082 | 6.2089 | 7.1544 | 6.4417 | | 2.5326 | 4.3644 | 5.1206 | H18 | 4.9010 | 3.3881 | 5.2509 | 3.8770 | 6.0499 | 7.2813 | 7.5156 | 6.6199 | 2.1893 | 1.0980 | 2.1615 | 3.4661 | 6.2089 | 8.2229 | 8.5850 | 7.1544 | 2.5326 | | 2.4673 | 4.3644 | H19 | 5.2509 | 3.8770 | 4.9010 | 3.3881 | 6.6199 | 7.5156 | 7.2813 | 6.0499 | 3.4661 | 2.1615 | 1.0980 | 2.1893 | 7.1544 | 8.5850 | 8.2229 | 6.2089 | 4.3644 | 2.4673 | | 2.5326 | H20 | 4.2591 | 3.4899 | 3.2814 | 2.1925 | 5.5954 | 6.0583 | 5.4322 | 4.0401 | 4.0237 | 3.4752 | 2.2097 | 1.0972 | 6.4417 | 7.1544 | 6.2089 | 3.9082 | 5.1206 | 4.3644 | 2.5326 | |
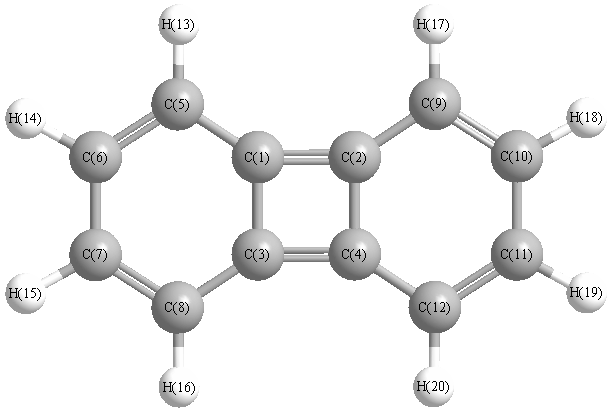 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
C4 |
90.000 |
|
C1 |
C2 |
C9 |
147.605 |
| C1 |
C3 |
C4 |
90.000 |
|
C1 |
C3 |
C8 |
122.395 |
| C1 |
C5 |
C6 |
115.578 |
|
C1 |
C5 |
H13 |
123.437 |
| C2 |
C1 |
C3 |
90.000 |
|
C2 |
C1 |
C5 |
147.605 |
| C2 |
C4 |
C3 |
90.000 |
|
C2 |
C4 |
C12 |
122.395 |
| C2 |
C9 |
C10 |
115.578 |
|
C2 |
C9 |
H17 |
123.437 |
| C3 |
C1 |
C5 |
122.395 |
|
C3 |
C4 |
C12 |
147.605 |
| C3 |
C8 |
C7 |
115.578 |
|
C3 |
C8 |
H16 |
123.437 |
| C4 |
C2 |
C9 |
122.395 |
|
C4 |
C3 |
C8 |
147.605 |
| C4 |
C12 |
C11 |
115.578 |
|
C4 |
C12 |
H20 |
123.437 |
| C5 |
C6 |
C7 |
122.027 |
|
C5 |
C6 |
H14 |
119.039 |
| C6 |
C5 |
H13 |
120.985 |
|
C6 |
C7 |
C8 |
122.027 |
| C6 |
C7 |
H15 |
118.934 |
|
C7 |
C6 |
H14 |
118.934 |
| C7 |
C8 |
H16 |
120.985 |
|
C8 |
C7 |
H15 |
119.039 |
| C9 |
C10 |
C11 |
122.027 |
|
C9 |
C10 |
H18 |
119.039 |
| C10 |
C9 |
H17 |
120.985 |
|
C10 |
C11 |
C12 |
122.027 |
| C10 |
C11 |
H19 |
118.934 |
|
C11 |
C10 |
H18 |
118.934 |
| C11 |
C12 |
H20 |
120.985 |
|
C12 |
C11 |
H19 |
119.039 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability