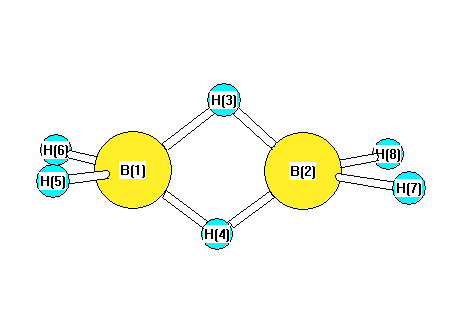
Vibrational Frequencies calculated at CCSD/aug-cc-pVDZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
Ag |
2615 |
2520 |
|
|
|
|
| 2 |
Ag |
2171 |
2092 |
|
|
|
|
| 3 |
Ag |
1200 |
1156 |
|
|
|
|
| 4 |
Ag |
807 |
778 |
|
|
|
|
| 5 |
Au |
837 |
807 |
|
|
|
|
| 6 |
B1g |
2691 |
2592 |
|
|
|
|
| 7 |
B1g |
929 |
895 |
|
|
|
|
| 8 |
B1u |
1988 |
1916 |
15.96 |
|
|
|
| 9 |
B1u |
990 |
953 |
26.02 |
|
|
|
| 10 |
B2g |
1880 |
1811 |
|
|
|
|
| 11 |
B2g |
880 |
848 |
|
|
|
|
| 12 |
B2u |
2707 |
2608 |
182.01 |
|
|
|
| 13 |
B2u |
977 |
941 |
2.34 |
|
|
|
| 14 |
B2u |
373 |
359 |
15.28 |
|
|
|
| 15 |
B3g |
1065 |
1026 |
|
|
|
|
| 16 |
B3u |
2599 |
2504 |
148.72 |
|
|
|
| 17 |
B3u |
1750 |
1686 |
550.40 |
|
|
|
| 18 |
B3u |
1184 |
1140 |
89.95 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 13820.8 cm
-1
Scaled (by 0.9634) Zero Point Vibrational Energy (zpe) 13314.9 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.