Jump to
S1C2
Vibrational Frequencies calculated at CCSD/cc-pVDZ
Geometric Data calculated at CCSD/cc-pVDZ
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Jump to
S1C1
Energy calculated at CCSD/cc-pVDZ
| | hartrees |
|---|
| Energy at 0K | -260.388621 |
| Energy at 298.15K | -260.393677 |
| HF Energy | -259.663244 |
| Nuclear repulsion energy | 127.235177 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at CCSD/cc-pVDZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3542 |
3355 |
23.71 |
|
|
|
| 2 |
A' |
1629 |
1543 |
53.70 |
|
|
|
| 3 |
A' |
1462 |
1385 |
186.47 |
|
|
|
| 4 |
A' |
1058 |
1002 |
22.55 |
|
|
|
| 5 |
A' |
878 |
832 |
210.71 |
|
|
|
| 6 |
A' |
751 |
712 |
56.52 |
|
|
|
| 7 |
A' |
677 |
641 |
27.79 |
|
|
|
| 8 |
A" |
3672 |
3478 |
39.20 |
|
|
|
| 9 |
A" |
1770 |
1677 |
318.44 |
|
|
|
| 10 |
A" |
1283 |
1215 |
62.15 |
|
|
|
| 11 |
A" |
576 |
546 |
1.98 |
|
|
|
| 12 |
A" |
386 |
365 |
32.01 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 8841.9 cm
-1
Scaled (by 0.9473) Zero Point Vibrational Energy (zpe) 8375.9 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at CCSD/cc-pVDZ
Point Group is Cs
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| N1 |
0.084 |
-1.255 |
0.000 |
| N2 |
0.007 |
0.148 |
0.000 |
| O3 |
0.007 |
0.682 |
1.091 |
| O4 |
0.007 |
0.682 |
-1.091 |
| H5 |
-0.378 |
-1.583 |
-0.848 |
| H6 |
-0.378 |
-1.583 |
0.848 |
Atom - Atom Distances (Å)
| |
N1 |
N2 |
O3 |
O4 |
H5 |
H6 |
| N1 | | 1.4048 | 2.2243 | 2.2243 | 1.0207 | 1.0207 |
N2 | 1.4048 | | 1.2147 | 1.2147 | 1.9664 | 1.9664 | O3 | 2.2243 | 1.2147 | | 2.1819 | 3.0071 | 2.3110 | O4 | 2.2243 | 1.2147 | 2.1819 | | 2.3110 | 3.0071 | H5 | 1.0207 | 1.9664 | 3.0071 | 2.3110 | | 1.6968 | H6 | 1.0207 | 1.9664 | 2.3110 | 3.0071 | 1.6968 | |
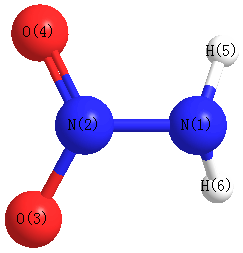 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| N1 |
N2 |
O3 |
116.043 |
|
N1 |
N2 |
O4 |
116.043 |
| N2 |
N1 |
H5 |
107.267 |
|
N2 |
N1 |
H6 |
107.267 |
| O3 |
N2 |
O4 |
127.829 |
|
H5 |
N1 |
H6 |
112.443 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability