Jump to
S1C2
Energy calculated at CCSD(T)/aug-cc-pVDZ
| | hartrees |
|---|
| Energy at 0K | -116.933696 |
| Energy at 298.15K | -116.937930 |
| HF Energy | -116.482280 |
| Nuclear repulsion energy | 64.155964 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at CCSD(T)/aug-cc-pVDZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A1 |
3250 |
3128 |
|
|
|
|
| 2 |
A1 |
3156 |
3038 |
|
|
|
|
| 3 |
A1 |
3143 |
3025 |
|
|
|
|
| 4 |
A1 |
1513 |
1457 |
|
|
|
|
| 5 |
A1 |
1260 |
1213 |
|
|
|
|
| 6 |
A1 |
1026 |
988 |
|
|
|
|
| 7 |
A1 |
420 |
405 |
|
|
|
|
| 8 |
A2 |
778 |
749 |
|
|
|
|
| 9 |
A2 |
548 |
527 |
|
|
|
|
| 10 |
B1 |
984 |
947 |
|
|
|
|
| 11 |
B1 |
793 |
763 |
|
|
|
|
| 12 |
B1 |
525 |
506 |
|
|
|
|
| 13 |
B2 |
3246 |
3125 |
|
|
|
|
| 14 |
B2 |
3139 |
3022 |
|
|
|
|
| 15 |
B2 |
1504 |
1448 |
|
|
|
|
| 16 |
B2 |
1405 |
1353 |
|
|
|
|
| 17 |
B2 |
1184 |
1139 |
|
|
|
|
| 18 |
B2 |
922 |
888 |
|
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 14398.9 cm
-1
Scaled (by 0.9625) Zero Point Vibrational Energy (zpe) 13858.9 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at CCSD(T)/aug-cc-pVDZ
Point Group is C2v
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.000 |
0.000 |
0.455 |
| H2 |
0.000 |
0.000 |
1.555 |
| C3 |
0.000 |
1.240 |
-0.201 |
| C4 |
0.000 |
-1.240 |
-0.201 |
| H5 |
0.000 |
2.181 |
0.359 |
| H6 |
0.000 |
-2.181 |
0.359 |
| H7 |
0.000 |
1.295 |
-1.297 |
| H8 |
0.000 |
-1.295 |
-1.297 |
Atom - Atom Distances (Å)
| |
C1 |
H2 |
C3 |
C4 |
H5 |
H6 |
H7 |
H8 |
| C1 | | 1.0995 | 1.4030 | 1.4030 | 2.1827 | 2.1827 | 2.1787 | 2.1787 |
H2 | 1.0995 | | 2.1496 | 2.1496 | 2.4870 | 2.4870 | 3.1318 | 3.1318 | C3 | 1.4030 | 2.1496 | | 2.4798 | 1.0946 | 3.4659 | 1.0970 | 2.7615 | C4 | 1.4030 | 2.1496 | 2.4798 | | 3.4659 | 1.0946 | 2.7615 | 1.0970 | H5 | 2.1827 | 2.4870 | 1.0946 | 3.4659 | | 4.3611 | 1.8774 | 3.8497 | H6 | 2.1827 | 2.4870 | 3.4659 | 1.0946 | 4.3611 | | 3.8497 | 1.8774 | H7 | 2.1787 | 3.1318 | 1.0970 | 2.7615 | 1.8774 | 3.8497 | | 2.5900 | H8 | 2.1787 | 3.1318 | 2.7615 | 1.0970 | 3.8497 | 1.8774 | 2.5900 | |
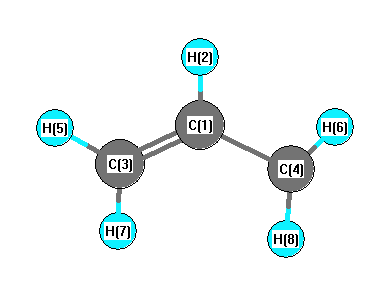 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C3 |
H5 |
121.340 |
|
C1 |
C3 |
H7 |
120.777 |
| C1 |
C4 |
H6 |
121.340 |
|
C1 |
C4 |
H8 |
120.777 |
| H2 |
C1 |
C3 |
117.899 |
|
H2 |
C1 |
C4 |
117.899 |
| C3 |
C1 |
C4 |
124.203 |
|
H5 |
C3 |
H7 |
117.883 |
| H6 |
C4 |
H8 |
117.883 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Jump to
S1C1
Energy calculated at CCSD(T)/aug-cc-pVDZ
| | hartrees |
|---|
| Energy at 0K | -116.933696 |
| Energy at 298.15K | -116.937893 |
| HF Energy | -116.482276 |
| Nuclear repulsion energy | 64.150425 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at CCSD(T)/aug-cc-pVDZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3248 |
3126 |
|
|
|
|
| 2 |
A' |
3244 |
3122 |
|
|
|
|
| 3 |
A' |
3153 |
3035 |
|
|
|
|
| 4 |
A' |
3140 |
3022 |
|
|
|
|
| 5 |
A' |
3136 |
3018 |
|
|
|
|
| 6 |
A' |
1508 |
1452 |
|
|
|
|
| 7 |
A' |
1500 |
1444 |
|
|
|
|
| 8 |
A' |
1403 |
1350 |
|
|
|
|
| 9 |
A' |
1257 |
1209 |
|
|
|
|
| 10 |
A' |
1179 |
1135 |
|
|
|
|
| 11 |
A' |
1023 |
984 |
|
|
|
|
| 12 |
A' |
913 |
879 |
|
|
|
|
| 13 |
A' |
411 |
396 |
|
|
|
|
| 14 |
A" |
977 |
940 |
|
|
|
|
| 15 |
A" |
791 |
762 |
|
|
|
|
| 16 |
A" |
768 |
739 |
|
|
|
|
| 17 |
A" |
530 |
510 |
|
|
|
|
| 18 |
A" |
516 |
497 |
|
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 14348.6 cm
-1
Scaled (by 0.9625) Zero Point Vibrational Energy (zpe) 13810.5 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at CCSD(T)/aug-cc-pVDZ
Point Group is Cs
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.000 |
0.455 |
0.000 |
| H2 |
0.001 |
1.555 |
0.000 |
| C3 |
-1.240 |
-0.201 |
0.000 |
| C4 |
1.240 |
-0.201 |
0.000 |
| H5 |
-2.181 |
0.360 |
0.000 |
| H6 |
2.180 |
0.360 |
0.000 |
| H7 |
-1.296 |
-1.296 |
0.000 |
| H8 |
1.297 |
-1.297 |
0.000 |
Atom - Atom Distances (Å)
| |
C1 |
H2 |
C3 |
C4 |
H5 |
H6 |
H7 |
H8 |
| C1 | | 1.0996 | 1.4029 | 1.4030 | 2.1827 | 2.1821 | 2.1787 | 2.1796 |
H2 | 1.0996 | | 2.1497 | 2.1494 | 2.4871 | 2.4855 | 3.1320 | 3.1322 | C3 | 1.4029 | 2.1497 | | 2.4801 | 1.0949 | 3.4658 | 1.0969 | 2.7634 | C4 | 1.4030 | 2.1494 | 2.4801 | | 3.4663 | 1.0947 | 2.7621 | 1.0969 | H5 | 2.1827 | 2.4871 | 1.0949 | 3.4663 | | 4.3606 | 1.8777 | 3.8517 | H6 | 2.1821 | 2.4855 | 3.4658 | 1.0947 | 4.3606 | | 3.8502 | 1.8773 | H7 | 2.1787 | 3.1320 | 1.0969 | 2.7621 | 1.8777 | 3.8502 | | 2.5925 | H8 | 2.1796 | 3.1322 | 2.7634 | 1.0969 | 3.8517 | 1.8773 | 2.5925 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C3 |
H5 |
121.322 |
|
C1 |
C3 |
H7 |
120.788 |
| C1 |
C4 |
H6 |
121.271 |
|
C1 |
C4 |
H8 |
120.860 |
| H2 |
C1 |
C3 |
117.909 |
|
H2 |
C1 |
C4 |
117.867 |
| C3 |
C1 |
C4 |
124.224 |
|
H5 |
C3 |
H7 |
117.890 |
| H6 |
C4 |
H8 |
117.869 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability