All results from a given calculation for PF3CH2 (phosphorane, trifluoromethylene-)
using model chemistry: CCSD(T)/aug-cc-pVTZ
19 10 17 12 22
States and conformations
| State |
Conformation |
minimum conformation |
conformer description |
state description |
| 1 |
1 |
yes |
CS |
1A' |
Energy calculated at CCSD(T)/aug-cc-pVTZ
| | hartrees |
|---|
| Energy at 0K | -679.448434 |
| Energy at 298.15K | |
| HF Energy | -678.299205 |
| Nuclear repulsion energy | 274.650492 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at CCSD(T)/aug-cc-pVTZ
Geometric Data calculated at CCSD(T)/aug-cc-pVTZ
Point Group is Cs
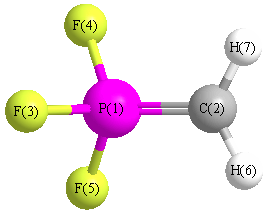
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| P1 |
-0.022 |
0.117 |
0.000 |
| C2 |
-0.492 |
1.668 |
0.000 |
| F3 |
1.457 |
-0.405 |
0.000 |
| F4 |
-0.492 |
-0.696 |
1.231 |
| F5 |
-0.492 |
-0.696 |
-1.231 |
| H6 |
-0.490 |
2.203 |
-0.938 |
| H7 |
-0.490 |
2.203 |
0.938 |
Atom - Atom Distances (Å)
| |
P1 |
C2 |
F3 |
F4 |
F5 |
H6 |
H7 |
| P1 | | 1.6204 | 1.5692 | 1.5484 | 1.5484 | 2.3340 | 2.3340 |
C2 | 1.6204 | | 2.8453 | 2.6652 | 2.6652 | 1.0797 | 1.0797 | F3 | 1.5692 | 2.8453 | | 2.3240 | 2.3240 | 3.3866 | 3.3866 | F4 | 1.5484 | 2.6652 | 2.3240 | | 2.4630 | 3.6204 | 2.9131 | F5 | 1.5484 | 2.6652 | 2.3240 | 2.4630 | | 2.9131 | 3.6204 | H6 | 2.3340 | 1.0797 | 3.3866 | 3.6204 | 2.9131 | | 1.8760 | H7 | 2.3340 | 1.0797 | 3.3866 | 2.9131 | 3.6204 | 1.8760 | |
 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| P1 |
C2 |
H6 |
118.254 |
|
P1 |
C2 |
H7 |
118.254 |
| C2 |
P1 |
F3 |
126.258 |
|
C2 |
P1 |
F4 |
114.495 |
| C2 |
P1 |
F5 |
114.495 |
|
F3 |
P1 |
F4 |
96.393 |
| F3 |
P1 |
F5 |
96.393 |
|
F4 |
P1 |
F5 |
105.377 |
| H6 |
C2 |
H7 |
120.634 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability