Jump to
S1C2
Energy calculated at CCSD(T)/6-31G(2df,p)
| | hartrees |
|---|
| Energy at 0K | -187.637546 |
| Energy at 298.15K | -187.641556 |
| HF Energy | -186.899134 |
| Nuclear repulsion energy | 101.523525 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at CCSD(T)/6-31G(2df,p)
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3654 |
3654 |
|
|
|
|
| 2 |
A |
3564 |
3564 |
|
|
|
|
| 3 |
A |
2392 |
2392 |
|
|
|
|
| 4 |
A |
1671 |
1671 |
|
|
|
|
| 5 |
A |
1222 |
1222 |
|
|
|
|
| 6 |
A |
820 |
820 |
|
|
|
|
| 7 |
A |
676 |
676 |
|
|
|
|
| 8 |
A |
388 |
388 |
|
|
|
|
| 9 |
A |
331 |
331 |
|
|
|
|
| 10 |
A |
201 |
201 |
|
|
|
|
| 11 |
B |
3654 |
3654 |
|
|
|
|
| 12 |
B |
3566 |
3566 |
|
|
|
|
| 13 |
B |
1669 |
1669 |
|
|
|
|
| 14 |
B |
1345 |
1345 |
|
|
|
|
| 15 |
B |
1222 |
1222 |
|
|
|
|
| 16 |
B |
739 |
739 |
|
|
|
|
| 17 |
B |
389 |
389 |
|
|
|
|
| 18 |
B |
202 |
202 |
|
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 13852.0 cm
-1
Scaled (by 1) Zero Point Vibrational Energy (zpe) 13852.0 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at CCSD(T)/6-31G(2df,p)
Point Group is C2
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.013 |
0.605 |
-0.003 |
| C2 |
-0.013 |
-0.605 |
-0.003 |
| N3 |
-0.013 |
1.972 |
-0.061 |
| N4 |
0.013 |
-1.972 |
-0.061 |
| H5 |
-0.294 |
2.399 |
0.812 |
| H6 |
0.867 |
2.368 |
-0.367 |
| H7 |
0.294 |
-2.399 |
0.812 |
| H8 |
-0.867 |
-2.368 |
-0.367 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
N3 |
N4 |
H5 |
H6 |
H7 |
H8 |
| C1 | | 1.2107 | 1.3681 | 2.5778 | 1.9940 | 1.9926 | 3.1257 | 3.1221 |
C2 | 1.2107 | | 2.5778 | 1.3681 | 3.1257 | 3.1221 | 1.9940 | 1.9926 | N3 | 1.3681 | 2.5778 | | 3.9439 | 1.0116 | 1.0117 | 4.4679 | 4.4341 | N4 | 2.5778 | 1.3681 | 3.9439 | | 4.4679 | 4.4341 | 1.0116 | 1.0117 | H5 | 1.9940 | 3.1257 | 1.0116 | 4.4679 | | 1.6542 | 4.8343 | 4.9443 | H6 | 1.9926 | 3.1221 | 1.0117 | 4.4341 | 1.6542 | | 4.9443 | 5.0440 | H7 | 3.1257 | 1.9940 | 4.4679 | 1.0116 | 4.8343 | 4.9443 | | 1.6542 | H8 | 3.1221 | 1.9926 | 4.4341 | 1.0117 | 4.9443 | 5.0440 | 1.6542 | |
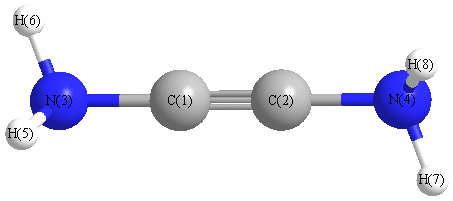 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
N4 |
176.706 |
|
C1 |
N3 |
H5 |
112.992 |
| C1 |
N3 |
H6 |
112.851 |
|
C2 |
C1 |
N3 |
176.706 |
| C2 |
N4 |
H7 |
112.992 |
|
C2 |
N4 |
H8 |
112.851 |
| H5 |
N3 |
H6 |
109.690 |
|
H7 |
N4 |
H8 |
109.690 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Jump to
S1C1
Energy calculated at CCSD(T)/6-31G(2df,p)
| | hartrees |
|---|
| Energy at 0K | -187.631897 |
| Energy at 298.15K | |
| HF Energy | -186.896062 |
| Nuclear repulsion energy | 102.172388 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at CCSD(T)/6-31G(2df,p)
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A1 |
2378 |
2378 |
|
|
|
|
| 2 |
A1 |
840 |
840 |
|
|
|
|
| 3 |
B1 |
508 |
508 |
|
|
|
|
| 4 |
B2 |
1426 |
1426 |
|
|
|
|
| 5 |
B2 |
409 |
409 |
|
|
|
|
| 6 |
B2 |
409 |
409 |
|
|
|
|
| 7 |
B2 |
160 |
160 |
|
|
|
|
| 8 |
B2 |
158 |
158 |
|
|
|
|
| 9 |
B2 |
516i |
516i |
|
|
|
|
| 9 |
B2 |
516i |
516i |
|
|
|
|
| 10 |
E |
3784 |
3784 |
|
|
|
|
| 10 |
E |
3784 |
3784 |
|
|
|
|
| 11 |
E |
3674 |
3674 |
|
|
|
|
| 11 |
E |
3668 |
3668 |
|
|
|
|
| 12 |
E |
1659 |
1659 |
|
|
|
|
| 12 |
E |
1652 |
1652 |
|
|
|
|
| 13 |
E |
1144 |
1144 |
|
|
|
|
| 13 |
E |
1144 |
1144 |
|
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 12880.6 cm
-1
Scaled (by 1) Zero Point Vibrational Energy (zpe) 12880.6 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at CCSD(T)/6-31G(2df,p)
Point Group is D2d
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.000 |
0.000 |
0.608 |
| C2 |
0.000 |
0.000 |
-0.608 |
| N3 |
0.000 |
0.000 |
1.948 |
| N4 |
0.000 |
0.000 |
-1.948 |
| H5 |
0.000 |
0.858 |
2.467 |
| H6 |
0.000 |
-0.858 |
2.467 |
| H7 |
0.858 |
0.000 |
-2.467 |
| H8 |
-0.858 |
0.000 |
-2.467 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
N3 |
N4 |
H5 |
H6 |
H7 |
H8 |
| C1 | | 1.2150 | 1.3409 | 2.5560 | 2.0474 | 2.0474 | 3.1915 | 3.1915 |
C2 | 1.2150 | | 2.5560 | 1.3409 | 3.1915 | 3.1915 | 2.0474 | 2.0474 | N3 | 1.3409 | 2.5560 | | 3.8969 | 1.0020 | 1.0020 | 4.4976 | 4.4976 | N4 | 2.5560 | 1.3409 | 3.8969 | | 4.4976 | 4.4976 | 1.0020 | 1.0020 | H5 | 2.0474 | 3.1915 | 1.0020 | 4.4976 | | 1.7153 | 5.0801 | 5.0801 | H6 | 2.0474 | 3.1915 | 1.0020 | 4.4976 | 1.7153 | | 5.0801 | 5.0801 | H7 | 3.1915 | 2.0474 | 4.4976 | 1.0020 | 5.0801 | 5.0801 | | 1.7153 | H8 | 3.1915 | 2.0474 | 4.4976 | 1.0020 | 5.0801 | 5.0801 | 1.7153 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
N4 |
180.000 |
|
C1 |
N3 |
H5 |
121.139 |
| C1 |
N3 |
H6 |
121.139 |
|
C2 |
C1 |
N3 |
180.000 |
| C2 |
N4 |
H7 |
121.139 |
|
C2 |
N4 |
H8 |
121.139 |
| H5 |
N3 |
H6 |
117.722 |
|
H7 |
N4 |
H8 |
117.722 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability