Jump to
S1C2
S1C3
Energy calculated at CCSD(T)/cc-pVQZ
| | hartrees |
|---|
| Energy at 0K | -100.202297 |
| Energy at 298.15K | -100.201345 |
| HF Energy | -99.815337 |
| Nuclear repulsion energy | 27.528824 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Geometric Data calculated at CCSD(T)/cc-pVQZ
Point Group is C∞v
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| Li1 |
0.000 |
0.000 |
-2.079 |
| C2 |
0.000 |
0.000 |
-0.150 |
| N3 |
0.000 |
0.000 |
1.020 |
Atom - Atom Distances (Å)
| |
Li1 |
C2 |
N3 |
| Li1 | | 1.9287 | 3.0986 |
C2 | 1.9287 | | 1.1699 | N3 | 3.0986 | 1.1699 | |
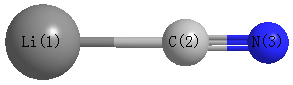 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| Li1 |
C2 |
N3 |
180.000 |
|
Li1 |
N3 |
C2 |
0.000 |
| C2 |
Li1 |
N3 |
0.000 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Jump to
S1C1
S1C3
Energy calculated at CCSD(T)/cc-pVQZ
| | hartrees |
|---|
| Energy at 0K | -100.205770 |
| Energy at 298.15K | -100.205013 |
| HF Energy | -99.821514 |
| Nuclear repulsion energy | 29.214851 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Geometric Data calculated at CCSD(T)/cc-pVQZ
Point Group is Cs
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| Li1 |
1.422 |
-0.621 |
0.000 |
| C2 |
-0.711 |
-0.365 |
0.000 |
| N3 |
0.000 |
0.579 |
0.000 |
Atom - Atom Distances (Å)
| |
Li1 |
C2 |
N3 |
| Li1 | | 2.1485 | 1.8609 |
C2 | 2.1485 | | 1.1817 | N3 | 1.8609 | 1.1817 | |
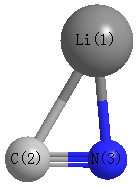 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| Li1 |
C2 |
N3 |
59.859 |
|
Li1 |
N3 |
C2 |
86.830 |
| C2 |
Li1 |
N3 |
33.311 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Jump to
S1C1
S1C2
Energy calculated at CCSD(T)/cc-pVQZ
| | hartrees |
|---|
| Energy at 0K | -100.206036 |
| Energy at 298.15K | -100.204901 |
| HF Energy | -99.826004 |
| Nuclear repulsion energy | 28.297595 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Geometric Data calculated at CCSD(T)/cc-pVQZ
Point Group is C∞v
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| Li1 |
0.000 |
0.000 |
1.890 |
| C2 |
0.000 |
0.000 |
-1.071 |
| N3 |
0.000 |
0.000 |
0.108 |
Atom - Atom Distances (Å)
| |
Li1 |
C2 |
N3 |
| Li1 | | 2.9612 | 1.7814 |
C2 | 2.9612 | | 1.1798 | N3 | 1.7814 | 1.1798 | |
 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| Li1 |
C2 |
N3 |
0.000 |
|
Li1 |
N3 |
C2 |
180.000 |
| C2 |
Li1 |
N3 |
0.000 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability