Jump to
S1C2
Energy calculated at BLYP/6-31G*
| | hartrees |
|---|
| Energy at 0K | -196.401113 |
| Energy at 298.15K | -196.411139 |
| HF Energy | -196.401113 |
| Nuclear repulsion energy | 168.130867 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at BLYP/6-31G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3146 |
3121 |
27.60 |
|
|
|
| 2 |
A |
3071 |
3046 |
9.34 |
|
|
|
| 3 |
A |
3045 |
3021 |
38.70 |
|
|
|
| 4 |
A |
3029 |
3004 |
43.68 |
|
|
|
| 5 |
A |
3024 |
3000 |
62.61 |
|
|
|
| 6 |
A |
2989 |
2965 |
24.08 |
|
|
|
| 7 |
A |
2971 |
2947 |
18.10 |
|
|
|
| 8 |
A |
2963 |
2939 |
32.94 |
|
|
|
| 9 |
A |
2950 |
2926 |
26.22 |
|
|
|
| 10 |
A |
2922 |
2898 |
26.59 |
|
|
|
| 11 |
A |
1671 |
1657 |
9.06 |
|
|
|
| 12 |
A |
1503 |
1490 |
2.54 |
|
|
|
| 13 |
A |
1492 |
1480 |
4.97 |
|
|
|
| 14 |
A |
1486 |
1474 |
0.35 |
|
|
|
| 15 |
A |
1472 |
1460 |
1.64 |
|
|
|
| 16 |
A |
1438 |
1426 |
0.67 |
|
|
|
| 17 |
A |
1400 |
1389 |
0.42 |
|
|
|
| 18 |
A |
1357 |
1346 |
1.67 |
|
|
|
| 19 |
A |
1310 |
1300 |
0.59 |
|
|
|
| 20 |
A |
1302 |
1291 |
0.61 |
|
|
|
| 21 |
A |
1278 |
1268 |
8.49 |
|
|
|
| 22 |
A |
1248 |
1238 |
0.11 |
|
|
|
| 23 |
A |
1175 |
1166 |
0.37 |
|
|
|
| 24 |
A |
1088 |
1080 |
0.67 |
|
|
|
| 25 |
A |
1024 |
1016 |
6.22 |
|
|
|
| 26 |
A |
1007 |
999 |
1.68 |
|
|
|
| 27 |
A |
1001 |
993 |
7.47 |
|
|
|
| 28 |
A |
928 |
921 |
0.33 |
|
|
|
| 29 |
A |
893 |
886 |
31.03 |
|
|
|
| 30 |
A |
868 |
861 |
5.81 |
|
|
|
| 31 |
A |
860 |
853 |
0.14 |
|
|
|
| 32 |
A |
739 |
734 |
2.46 |
|
|
|
| 33 |
A |
623 |
618 |
6.48 |
|
|
|
| 34 |
A |
425 |
422 |
1.42 |
|
|
|
| 35 |
A |
372 |
369 |
0.08 |
|
|
|
| 36 |
A |
246 |
244 |
0.02 |
|
|
|
| 37 |
A |
225 |
224 |
0.07 |
|
|
|
| 38 |
A |
103 |
103 |
0.03 |
|
|
|
| 39 |
A |
95 |
95 |
0.05 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 29370.2 cm
-1
Scaled (by 0.9919) Zero Point Vibrational Energy (zpe) 29132.3 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at BLYP/6-31G*
Point Group is C1
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
2.509 |
-0.218 |
-0.306 |
| C2 |
1.433 |
0.343 |
0.272 |
| C3 |
0.076 |
-0.314 |
0.438 |
| C4 |
-1.068 |
0.451 |
-0.284 |
| C5 |
-2.455 |
-0.199 |
-0.081 |
| H6 |
3.460 |
0.316 |
-0.403 |
| H7 |
2.484 |
-1.238 |
-0.706 |
| H8 |
1.508 |
1.371 |
0.656 |
| H9 |
0.120 |
-1.353 |
0.067 |
| H10 |
-0.171 |
-0.375 |
1.517 |
| H11 |
-1.093 |
1.495 |
0.079 |
| H12 |
-0.836 |
0.506 |
-1.362 |
| H13 |
-3.244 |
0.365 |
-0.606 |
| H14 |
-2.472 |
-1.233 |
-0.464 |
| H15 |
-2.726 |
-0.236 |
0.988 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
C3 |
C4 |
C5 |
H6 |
H7 |
H8 |
H9 |
H10 |
H11 |
H12 |
H13 |
H14 |
H15 |
| C1 | | 1.3453 | 2.5468 | 3.6393 | 4.9695 | 1.0944 | 1.0958 | 2.1103 | 2.6716 | 3.2455 | 4.0079 | 3.5817 | 5.7905 | 5.0863 | 5.3935 |
C2 | 1.3453 | | 1.5166 | 2.5637 | 3.9409 | 2.1371 | 2.1366 | 1.0993 | 2.1550 | 2.1533 | 2.7827 | 2.8005 | 4.7582 | 4.2749 | 4.2597 | C3 | 2.5468 | 1.5166 | | 1.5534 | 2.5859 | 3.5435 | 2.8225 | 2.2219 | 1.1049 | 1.1081 | 2.1833 | 2.1781 | 3.5456 | 2.8551 | 2.8566 | C4 | 3.6393 | 2.5637 | 1.5534 | | 1.5449 | 4.5315 | 3.9559 | 2.8925 | 2.1881 | 2.1744 | 1.1058 | 1.1046 | 2.2014 | 2.2000 | 2.2003 | C5 | 4.9695 | 3.9409 | 2.5859 | 1.5449 | | 5.9460 | 5.0862 | 4.3259 | 2.8258 | 2.7927 | 2.1788 | 2.1820 | 1.1029 | 1.1037 | 1.1038 | H6 | 1.0944 | 2.1371 | 3.5435 | 4.5315 | 5.9460 | | 1.8595 | 2.4585 | 3.7632 | 4.1649 | 4.7283 | 4.4057 | 6.7072 | 6.1313 | 6.3648 | H7 | 1.0958 | 2.1366 | 2.8225 | 3.9559 | 5.0862 | 1.8595 | | 3.1009 | 2.4903 | 3.5692 | 4.5701 | 3.8072 | 5.9493 | 4.9624 | 5.5703 | H8 | 2.1103 | 1.0993 | 2.2219 | 2.8925 | 4.3259 | 2.4585 | 3.1009 | | 3.1137 | 2.5707 | 2.6678 | 3.2118 | 5.0187 | 4.8867 | 4.5415 | H9 | 2.6716 | 2.1550 | 1.1049 | 2.1881 | 2.8258 | 3.7632 | 2.4903 | 3.1137 | | 1.7732 | 3.0959 | 2.5322 | 3.8368 | 2.6486 | 3.1935 | H10 | 3.2455 | 2.1533 | 1.1081 | 2.1744 | 2.7927 | 4.1649 | 3.5692 | 2.5707 | 1.7732 | | 2.5328 | 3.0833 | 3.8074 | 3.1553 | 2.6129 | H11 | 4.0079 | 2.7827 | 2.1833 | 1.1058 | 2.1788 | 4.7283 | 4.5701 | 2.6678 | 3.0959 | 2.5328 | | 1.7666 | 2.5239 | 3.1047 | 2.5479 | H12 | 3.5817 | 2.8005 | 2.1781 | 1.1046 | 2.1820 | 4.4057 | 3.8072 | 3.2118 | 2.5322 | 3.0833 | 1.7666 | | 2.5281 | 2.5513 | 3.1068 | H13 | 5.7905 | 4.7582 | 3.5456 | 2.2014 | 1.1029 | 6.7072 | 5.9493 | 5.0187 | 3.8368 | 3.8074 | 2.5239 | 2.5281 | | 1.7807 | 1.7812 | H14 | 5.0863 | 4.2749 | 2.8551 | 2.2000 | 1.1037 | 6.1313 | 4.9624 | 4.8867 | 2.6486 | 3.1553 | 3.1047 | 2.5513 | 1.7807 | | 1.7802 | H15 | 5.3935 | 4.2597 | 2.8566 | 2.2003 | 1.1038 | 6.3648 | 5.5703 | 4.5415 | 3.1935 | 2.6129 | 2.5479 | 3.1068 | 1.7812 | 1.7802 | |
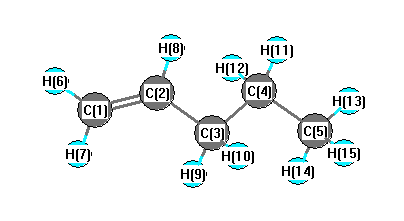 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
C3 |
125.618 |
|
C1 |
C2 |
H8 |
119.024 |
| C2 |
C1 |
H6 |
121.980 |
|
C2 |
C1 |
H7 |
121.820 |
| C2 |
C3 |
C4 |
113.241 |
|
C2 |
C3 |
H9 |
109.572 |
| C2 |
C3 |
H10 |
109.258 |
|
C3 |
C2 |
H8 |
115.353 |
| C3 |
C4 |
C5 |
113.149 |
|
C3 |
C4 |
H11 |
109.220 |
| C3 |
C4 |
H12 |
108.887 |
|
C4 |
C3 |
H9 |
109.638 |
| C4 |
C3 |
H10 |
108.404 |
|
C4 |
C5 |
H13 |
111.395 |
| C4 |
C5 |
H14 |
111.233 |
|
C4 |
C5 |
H15 |
111.252 |
| C5 |
C4 |
H11 |
109.454 |
|
C5 |
C4 |
H12 |
109.768 |
| H6 |
C1 |
H7 |
116.199 |
|
H9 |
C3 |
H10 |
106.498 |
| H11 |
C4 |
H12 |
106.120 |
|
H13 |
C5 |
H14 |
107.615 |
| H13 |
C5 |
H15 |
107.647 |
|
H14 |
C5 |
H15 |
107.506 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at BLYP/6-31G*
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.308 |
|
|
|
| 2 |
C |
-0.014 |
|
|
|
| 3 |
C |
-0.259 |
|
|
|
| 4 |
C |
-0.211 |
|
|
|
| 5 |
C |
-0.403 |
|
|
|
| 6 |
H |
0.118 |
|
|
|
| 7 |
H |
0.114 |
|
|
|
| 8 |
H |
0.098 |
|
|
|
| 9 |
H |
0.117 |
|
|
|
| 10 |
H |
0.122 |
|
|
|
| 11 |
H |
0.116 |
|
|
|
| 12 |
H |
0.124 |
|
|
|
| 13 |
H |
0.129 |
|
|
|
| 14 |
H |
0.129 |
|
|
|
| 15 |
H |
0.128 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
-0.364 |
0.080 |
0.072 |
0.379 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-33.530 |
0.161 |
-0.472 |
| y |
0.161 |
-32.523 |
0.932 |
| z |
-0.472 |
0.932 |
-33.863 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-0.337 |
0.161 |
-0.472 |
| y |
0.161 |
1.173 |
0.932 |
| z |
-0.472 |
0.932 |
-0.836 |
|
| Polar |
|---|
| 3z2-r2 | -1.673 |
|---|
| x2-y2 | -1.007 |
|---|
| xy | 0.161 |
|---|
| xz | -0.472 |
|---|
| yz | 0.932 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
10.760 |
-0.296 |
-0.946 |
| y |
-0.296 |
7.345 |
0.645 |
| z |
-0.946 |
0.645 |
6.356 |
<r2> (average value of r
2) Å
2
| <r2> |
182.088 |
| (<r2>)1/2 |
13.494 |
Jump to
S1C1
Energy calculated at BLYP/6-31G*
| | hartrees |
|---|
| Energy at 0K | -196.400649 |
| Energy at 298.15K | -196.410750 |
| HF Energy | -196.400649 |
| Nuclear repulsion energy | 171.335510 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at BLYP/6-31G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3146 |
3121 |
26.42 |
|
|
|
| 2 |
A |
3071 |
3046 |
9.02 |
|
|
|
| 3 |
A |
3041 |
3017 |
41.09 |
|
|
|
| 4 |
A |
3038 |
3013 |
39.43 |
|
|
|
| 5 |
A |
3025 |
3001 |
45.00 |
|
|
|
| 6 |
A |
2983 |
2959 |
40.42 |
|
|
|
| 7 |
A |
2973 |
2949 |
27.60 |
|
|
|
| 8 |
A |
2967 |
2942 |
24.11 |
|
|
|
| 9 |
A |
2947 |
2923 |
32.24 |
|
|
|
| 10 |
A |
2929 |
2905 |
30.56 |
|
|
|
| 11 |
A |
1670 |
1656 |
7.21 |
|
|
|
| 12 |
A |
1502 |
1490 |
1.92 |
|
|
|
| 13 |
A |
1493 |
1481 |
6.14 |
|
|
|
| 14 |
A |
1481 |
1469 |
0.18 |
|
|
|
| 15 |
A |
1471 |
1459 |
3.75 |
|
|
|
| 16 |
A |
1438 |
1427 |
0.60 |
|
|
|
| 17 |
A |
1403 |
1392 |
2.97 |
|
|
|
| 18 |
A |
1354 |
1343 |
9.54 |
|
|
|
| 19 |
A |
1334 |
1323 |
2.50 |
|
|
|
| 20 |
A |
1303 |
1292 |
1.63 |
|
|
|
| 21 |
A |
1269 |
1259 |
0.21 |
|
|
|
| 22 |
A |
1236 |
1226 |
1.77 |
|
|
|
| 23 |
A |
1171 |
1161 |
0.41 |
|
|
|
| 24 |
A |
1075 |
1067 |
4.81 |
|
|
|
| 25 |
A |
1038 |
1029 |
2.87 |
|
|
|
| 26 |
A |
1006 |
998 |
8.80 |
|
|
|
| 27 |
A |
995 |
987 |
1.63 |
|
|
|
| 28 |
A |
909 |
901 |
0.89 |
|
|
|
| 29 |
A |
894 |
887 |
32.96 |
|
|
|
| 30 |
A |
871 |
864 |
0.30 |
|
|
|
| 31 |
A |
838 |
831 |
1.73 |
|
|
|
| 32 |
A |
758 |
751 |
3.66 |
|
|
|
| 33 |
A |
625 |
620 |
6.12 |
|
|
|
| 34 |
A |
423 |
419 |
0.36 |
|
|
|
| 35 |
A |
379 |
376 |
1.61 |
|
|
|
| 36 |
A |
293 |
291 |
0.18 |
|
|
|
| 37 |
A |
218 |
217 |
0.17 |
|
|
|
| 38 |
A |
121 |
120 |
0.02 |
|
|
|
| 39 |
A |
92 |
91 |
0.06 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 29388.4 cm
-1
Scaled (by 0.9919) Zero Point Vibrational Energy (zpe) 29150.3 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at BLYP/6-31G*
Point Group is C1
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
-2.144 |
0.575 |
0.118 |
| C2 |
-1.207 |
-0.240 |
-0.400 |
| C3 |
-0.076 |
-0.896 |
0.369 |
| C4 |
1.340 |
-0.530 |
-0.162 |
| C5 |
1.715 |
0.956 |
0.025 |
| H6 |
-2.937 |
1.011 |
-0.498 |
| H7 |
-2.159 |
0.836 |
1.182 |
| H8 |
-1.236 |
-0.469 |
-1.476 |
| H9 |
-0.195 |
-1.996 |
0.309 |
| H10 |
-0.154 |
-0.629 |
1.439 |
| H11 |
2.084 |
-1.164 |
0.353 |
| H12 |
1.403 |
-0.795 |
-1.234 |
| H13 |
2.727 |
1.163 |
-0.359 |
| H14 |
1.010 |
1.619 |
-0.503 |
| H15 |
1.698 |
1.238 |
1.092 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
C3 |
C4 |
C5 |
H6 |
H7 |
H8 |
H9 |
H10 |
H11 |
H12 |
H13 |
H14 |
H15 |
| C1 | | 1.3455 | 2.5501 | 3.6655 | 3.8786 | 1.0945 | 1.0957 | 2.1102 | 3.2314 | 2.6753 | 4.5776 | 4.0355 | 4.9301 | 3.3795 | 4.0187 |
C2 | 1.3455 | | 1.5170 | 2.5745 | 3.1859 | 2.1373 | 2.1368 | 1.0997 | 2.1470 | 2.1549 | 3.5005 | 2.7958 | 4.1777 | 2.8950 | 3.5852 | C3 | 2.5501 | 1.5170 | | 1.5562 | 2.5998 | 3.5459 | 2.8278 | 2.2204 | 1.1075 | 1.1051 | 2.1769 | 2.1837 | 3.5543 | 2.8751 | 2.8686 | C4 | 3.6655 | 2.5745 | 1.5562 | | 1.5442 | 4.5584 | 3.9886 | 2.8922 | 2.1745 | 2.1911 | 1.1050 | 1.1060 | 2.1978 | 2.2006 | 2.1973 | C5 | 3.8786 | 3.1859 | 2.5998 | 1.5442 | | 4.6811 | 4.0441 | 3.6043 | 3.5278 | 2.8288 | 2.1769 | 2.1791 | 1.1029 | 1.1021 | 1.1039 | H6 | 1.0945 | 2.1373 | 3.5459 | 4.5584 | 4.6811 | | 1.8594 | 2.4579 | 4.1485 | 3.7668 | 5.5376 | 4.7582 | 5.6682 | 3.9931 | 4.9052 | H7 | 1.0957 | 2.1368 | 2.8278 | 3.9886 | 4.0441 | 1.8594 | | 3.1009 | 3.5542 | 2.4965 | 4.7627 | 4.6022 | 5.1339 | 3.6728 | 3.8785 | H8 | 2.1102 | 1.0997 | 2.2204 | 2.8922 | 3.6043 | 2.4579 | 3.1009 | | 2.5691 | 3.1129 | 3.8537 | 2.6702 | 4.4295 | 3.2170 | 4.2565 | H9 | 3.2314 | 2.1470 | 1.1075 | 2.1745 | 3.5278 | 4.1485 | 3.5542 | 2.5691 | | 1.7732 | 2.4271 | 2.5259 | 4.3554 | 3.8960 | 3.8286 | H10 | 2.6753 | 2.1549 | 1.1051 | 2.1911 | 2.8288 | 3.7668 | 2.4965 | 3.1129 | 1.7732 | | 2.5436 | 3.0971 | 3.8398 | 3.1901 | 2.6525 | H11 | 4.5776 | 3.5005 | 2.1769 | 1.1050 | 2.1769 | 5.5376 | 4.7627 | 3.8537 | 2.4271 | 2.5436 | | 1.7660 | 2.5169 | 3.1032 | 2.5427 | H12 | 4.0355 | 2.7958 | 2.1837 | 1.1060 | 2.1791 | 4.7582 | 4.6022 | 2.6702 | 2.5259 | 3.0971 | 1.7660 | | 2.5202 | 2.5524 | 3.1035 | H13 | 4.9301 | 4.1777 | 3.5543 | 2.1978 | 1.1029 | 5.6682 | 5.1339 | 4.4295 | 4.3554 | 3.8398 | 2.5169 | 2.5202 | | 1.7832 | 1.7813 | H14 | 3.3795 | 2.8950 | 2.8751 | 2.2006 | 1.1021 | 3.9931 | 3.6728 | 3.2170 | 3.8960 | 3.1901 | 3.1032 | 2.5524 | 1.7832 | | 1.7785 | H15 | 4.0187 | 3.5852 | 2.8686 | 2.1973 | 1.1039 | 4.9052 | 3.8785 | 4.2565 | 3.8286 | 2.6525 | 2.5427 | 3.1035 | 1.7813 | 1.7785 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
C3 |
125.856 |
|
C1 |
C2 |
H8 |
118.966 |
| C2 |
C1 |
H6 |
121.974 |
|
C2 |
C1 |
H7 |
121.829 |
| C2 |
C3 |
C4 |
113.796 |
|
C2 |
C3 |
H9 |
108.781 |
| C2 |
C3 |
H10 |
109.530 |
|
C3 |
C2 |
H8 |
115.177 |
| C3 |
C4 |
C5 |
113.972 |
|
C3 |
C4 |
H11 |
108.591 |
| C3 |
C4 |
H12 |
109.048 |
|
C4 |
C3 |
H9 |
108.262 |
| C4 |
C3 |
H10 |
109.676 |
|
C4 |
C5 |
H13 |
111.158 |
| C4 |
C5 |
H14 |
111.423 |
|
C4 |
C5 |
H15 |
111.053 |
| C5 |
C4 |
H11 |
109.397 |
|
C5 |
C4 |
H12 |
109.508 |
| H6 |
C1 |
H7 |
116.196 |
|
H9 |
C3 |
H10 |
106.531 |
| H11 |
C4 |
H12 |
106.019 |
|
H13 |
C5 |
H14 |
107.937 |
| H13 |
C5 |
H15 |
107.637 |
|
H14 |
C5 |
H15 |
107.454 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at BLYP/6-31G*
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.308 |
|
|
|
| 2 |
C |
-0.009 |
|
|
|
| 3 |
C |
-0.261 |
|
|
|
| 4 |
C |
-0.215 |
|
|
|
| 5 |
C |
-0.399 |
|
|
|
| 6 |
H |
0.118 |
|
|
|
| 7 |
H |
0.115 |
|
|
|
| 8 |
H |
0.098 |
|
|
|
| 9 |
H |
0.122 |
|
|
|
| 10 |
H |
0.117 |
|
|
|
| 11 |
H |
0.115 |
|
|
|
| 12 |
H |
0.116 |
|
|
|
| 13 |
H |
0.126 |
|
|
|
| 14 |
H |
0.139 |
|
|
|
| 15 |
H |
0.126 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.257 |
-0.221 |
-0.053 |
0.344 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-33.707 |
-1.032 |
0.338 |
| y |
-1.032 |
-33.725 |
0.376 |
| z |
0.338 |
0.376 |
-32.185 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-0.752 |
-1.032 |
0.338 |
| y |
-1.032 |
-0.779 |
0.376 |
| z |
0.338 |
0.376 |
1.532 |
|
| Polar |
|---|
| 3z2-r2 | 3.063 |
|---|
| x2-y2 | 0.018 |
|---|
| xy | -1.032 |
|---|
| xz | 0.338 |
|---|
| yz | 0.376 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
9.390 |
-1.448 |
-0.156 |
| y |
-1.448 |
7.389 |
0.450 |
| z |
-0.156 |
0.450 |
7.368 |
<r2> (average value of r
2) Å
2
| <r2> |
156.758 |
| (<r2>)1/2 |
12.520 |