Jump to
S1C2
S1C3
Energy calculated at BLYP/aug-cc-pVTZ
| | hartrees |
|---|
| Energy at 0K | -581.329019 |
| Energy at 298.15K | |
| HF Energy | -581.329019 |
| Nuclear repulsion energy | 78.311012 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at BLYP/aug-cc-pVTZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
Ag |
2196 |
2189 |
0.00 |
419.63 |
0.13 |
0.24 |
| 2 |
Ag |
917 |
914 |
0.00 |
20.66 |
0.17 |
0.29 |
| 3 |
Ag |
564 |
562 |
0.00 |
117.76 |
0.12 |
0.22 |
| 4 |
Au |
529 |
527 |
0.00 |
0.00 |
0.00 |
0.00 |
| 5 |
B1u |
2191 |
2184 |
60.36 |
0.00 |
0.00 |
0.00 |
| 6 |
B1u |
841 |
838 |
111.76 |
0.00 |
0.00 |
0.00 |
| 7 |
B2g |
276i |
275i |
0.00 |
23.20 |
0.75 |
0.86 |
| 8 |
B2u |
2231 |
2223 |
96.92 |
0.00 |
0.00 |
0.00 |
| 9 |
B2u |
345 |
344 |
17.98 |
0.00 |
0.00 |
0.00 |
| 10 |
B3g |
2219 |
2212 |
0.00 |
221.68 |
0.75 |
0.86 |
| 11 |
B3g |
581 |
579 |
0.00 |
4.37 |
0.75 |
0.86 |
| 12 |
B3u |
490 |
489 |
0.02 |
0.00 |
0.00 |
0.00 |
Unscaled Zero Point Vibrational Energy (zpe) 6413.9 cm
-1
Scaled (by 0.9966) Zero Point Vibrational Energy (zpe) 6392.1 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at BLYP/aug-cc-pVTZ
Point Group is D2h
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| Si1 |
0.000 |
0.000 |
1.077 |
| Si2 |
0.000 |
0.000 |
-1.077 |
| H3 |
0.000 |
1.255 |
1.868 |
| H4 |
0.000 |
-1.255 |
1.868 |
| H5 |
0.000 |
1.255 |
-1.868 |
| H6 |
0.000 |
-1.255 |
-1.868 |
Atom - Atom Distances (Å)
| |
Si1 |
Si2 |
H3 |
H4 |
H5 |
H6 |
| Si1 | | 2.1547 | 1.4833 | 1.4833 | 3.2018 | 3.2018 |
Si2 | 2.1547 | | 3.2018 | 3.2018 | 1.4833 | 1.4833 | H3 | 1.4833 | 3.2018 | | 2.5096 | 3.7366 | 4.5011 | H4 | 1.4833 | 3.2018 | 2.5096 | | 4.5011 | 3.7366 | H5 | 3.2018 | 1.4833 | 3.7366 | 4.5011 | | 2.5096 | H6 | 3.2018 | 1.4833 | 4.5011 | 3.7366 | 2.5096 | |
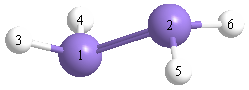 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| Si1 |
Si2 |
H5 |
122.225 |
|
Si1 |
Si2 |
H6 |
122.225 |
| Si2 |
Si1 |
H3 |
122.225 |
|
Si2 |
Si1 |
H4 |
122.225 |
| H3 |
Si1 |
H4 |
115.549 |
|
H5 |
Si2 |
H6 |
115.549 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at BLYP/aug-cc-pVTZ
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
Si |
0.317 |
|
|
|
| 2 |
Si |
0.317 |
|
|
|
| 3 |
H |
-0.158 |
|
|
|
| 4 |
H |
-0.158 |
|
|
|
| 5 |
H |
-0.158 |
|
|
|
| 6 |
H |
-0.158 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
0.000 |
0.000 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-31.955 |
0.000 |
0.000 |
| y |
0.000 |
-29.611 |
0.000 |
| z |
0.000 |
0.000 |
-28.001 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-3.149 |
0.000 |
0.000 |
| y |
0.000 |
0.367 |
0.000 |
| z |
0.000 |
0.000 |
2.782 |
|
| Polar |
|---|
| 3z2-r2 | 5.564 |
|---|
| x2-y2 | -2.344 |
|---|
| xy | 0.000 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
9.922 |
0.000 |
0.000 |
| y |
0.000 |
9.267 |
0.000 |
| z |
0.000 |
0.000 |
15.493 |
<r2> (average value of r
2) Å
2
| <r2> |
71.406 |
| (<r2>)1/2 |
8.450 |
Jump to
S1C1
S1C3
Energy calculated at BLYP/aug-cc-pVTZ
| | hartrees |
|---|
| Energy at 0K | -581.331006 |
| Energy at 298.15K | |
| HF Energy | -581.331006 |
| Nuclear repulsion energy | 77.357002 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at BLYP/aug-cc-pVTZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
Ag |
2152 |
2145 |
0.00 |
471.00 |
0.15 |
0.26 |
| 2 |
Ag |
925 |
922 |
0.00 |
21.30 |
0.25 |
0.40 |
| 3 |
Ag |
536 |
534 |
0.00 |
83.29 |
0.11 |
0.20 |
| 4 |
Ag |
355 |
354 |
0.00 |
16.04 |
0.46 |
0.63 |
| 5 |
Au |
2185 |
2177 |
142.74 |
0.00 |
0.00 |
0.00 |
| 6 |
Au |
499 |
497 |
0.01 |
0.00 |
0.00 |
0.00 |
| 7 |
Au |
333 |
332 |
15.73 |
0.00 |
0.00 |
0.00 |
| 8 |
Bg |
2172 |
2165 |
0.00 |
261.22 |
0.75 |
0.86 |
| 9 |
Bg |
596 |
594 |
0.00 |
3.98 |
0.75 |
0.86 |
| 10 |
Bu |
2150 |
2142 |
120.09 |
0.00 |
0.00 |
0.00 |
| 11 |
Bu |
901 |
898 |
175.67 |
0.00 |
0.00 |
0.00 |
| 12 |
Bu |
402 |
401 |
26.58 |
0.00 |
0.00 |
0.00 |
Unscaled Zero Point Vibrational Energy (zpe) 6602.7 cm
-1
Scaled (by 0.9966) Zero Point Vibrational Energy (zpe) 6580.3 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at BLYP/aug-cc-pVTZ
Point Group is C2h
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| Si1 |
0.000 |
1.098 |
0.000 |
| Si2 |
0.000 |
-1.098 |
0.000 |
| H3 |
0.477 |
1.796 |
1.229 |
| H4 |
0.477 |
1.796 |
-1.229 |
| H5 |
-0.477 |
-1.796 |
1.229 |
| H6 |
-0.477 |
-1.796 |
-1.229 |
Atom - Atom Distances (Å)
| |
Si1 |
Si2 |
H3 |
H4 |
H5 |
H6 |
| Si1 | | 2.1963 | 1.4919 | 1.4919 | 3.1805 | 3.1805 |
Si2 | 2.1963 | | 3.1805 | 3.1805 | 1.4919 | 1.4919 | H3 | 1.4919 | 3.1805 | | 2.4588 | 3.7165 | 4.4563 | H4 | 1.4919 | 3.1805 | 2.4588 | | 4.4563 | 3.7165 | H5 | 3.1805 | 1.4919 | 3.7165 | 4.4563 | | 2.4588 | H6 | 3.1805 | 1.4919 | 4.4563 | 3.7165 | 2.4588 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| Si1 |
Si2 |
H5 |
117.893 |
|
Si1 |
Si2 |
H6 |
117.893 |
| Si2 |
Si1 |
H3 |
117.893 |
|
Si2 |
Si1 |
H4 |
117.893 |
| H3 |
Si1 |
H4 |
110.988 |
|
H5 |
Si2 |
H6 |
110.988 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at BLYP/aug-cc-pVTZ
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
Si |
0.281 |
|
|
|
| 2 |
Si |
0.281 |
|
|
|
| 3 |
H |
-0.140 |
|
|
|
| 4 |
H |
-0.140 |
|
|
|
| 5 |
H |
-0.140 |
|
|
|
| 6 |
H |
-0.140 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
0.000 |
0.000 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-31.657 |
-0.567 |
0.000 |
| y |
-0.567 |
-28.298 |
0.000 |
| z |
0.000 |
0.000 |
-30.022 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-2.497 |
-0.567 |
0.000 |
| y |
-0.567 |
2.541 |
0.000 |
| z |
0.000 |
0.000 |
-0.045 |
|
| Polar |
|---|
| 3z2-r2 | -0.090 |
|---|
| x2-y2 | -3.359 |
|---|
| xy | -0.567 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
9.651 |
-0.414 |
0.000 |
| y |
-0.414 |
15.930 |
0.000 |
| z |
0.000 |
0.000 |
9.302 |
<r2> (average value of r
2) Å
2
| <r2> |
72.359 |
| (<r2>)1/2 |
8.506 |
Jump to
S1C1
S1C2
Energy calculated at BLYP/aug-cc-pVTZ
| | hartrees |
|---|
| Energy at 0K | -581.297413 |
| Energy at 298.15K | |
| HF Energy | -581.297413 |
| Nuclear repulsion energy | 72.040995 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at BLYP/aug-cc-pVTZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A1 |
2008 |
2001 |
349.39 |
288.90 |
0.27 |
0.42 |
| 2 |
A1 |
1564 |
1559 |
0.13 |
65.37 |
0.06 |
0.12 |
| 3 |
A1 |
854 |
851 |
20.77 |
81.84 |
0.18 |
0.31 |
| 4 |
A1 |
368 |
367 |
0.27 |
43.34 |
0.43 |
0.60 |
| 5 |
A1 |
362 |
361 |
0.77 |
11.74 |
0.05 |
0.10 |
| 6 |
A2 |
1294 |
1289 |
0.00 |
0.30 |
0.75 |
0.86 |
| 7 |
A2 |
608 |
606 |
0.00 |
0.27 |
0.75 |
0.86 |
| 8 |
B1 |
1213 |
1208 |
24.87 |
0.24 |
0.75 |
0.86 |
| 9 |
B1 |
789 |
786 |
15.13 |
3.80 |
0.75 |
0.86 |
| 10 |
B2 |
1983 |
1976 |
14.51 |
49.15 |
0.75 |
0.86 |
| 11 |
B2 |
1279 |
1275 |
915.74 |
36.66 |
0.75 |
0.86 |
| 12 |
B2 |
701 |
699 |
70.32 |
0.06 |
0.75 |
0.86 |
Unscaled Zero Point Vibrational Energy (zpe) 6511.6 cm
-1
Scaled (by 0.9966) Zero Point Vibrational Energy (zpe) 6489.4 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at BLYP/aug-cc-pVTZ
Point Group is C2v
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| Si1 |
0.000 |
1.340 |
-0.092 |
| Si2 |
0.000 |
-1.340 |
-0.092 |
| H3 |
-1.029 |
0.000 |
-0.150 |
| H4 |
1.029 |
0.000 |
-0.150 |
| H5 |
0.000 |
1.371 |
1.436 |
| H6 |
0.000 |
-1.371 |
1.436 |
Atom - Atom Distances (Å)
| |
Si1 |
Si2 |
H3 |
H4 |
H5 |
H6 |
| Si1 | | 2.6802 | 1.6907 | 1.6907 | 1.5277 | 3.1121 |
Si2 | 2.6802 | | 1.6907 | 1.6907 | 3.1121 | 1.5277 | H3 | 1.6907 | 1.6907 | | 2.0584 | 2.3357 | 2.3357 | H4 | 1.6907 | 1.6907 | 2.0584 | | 2.3357 | 2.3357 | H5 | 1.5277 | 3.1121 | 2.3357 | 2.3357 | | 2.7427 | H6 | 3.1121 | 1.5277 | 2.3357 | 2.3357 | 2.7427 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| Si1 |
Si2 |
H5 |
29.393 |
|
Si1 |
Si2 |
H6 |
91.172 |
| Si2 |
Si1 |
H3 |
37.569 |
|
Si2 |
Si1 |
H4 |
37.569 |
| H3 |
Si1 |
H4 |
74.995 |
|
H5 |
Si2 |
H6 |
61.780 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at BLYP/aug-cc-pVTZ
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
Si |
0.097 |
|
|
|
| 2 |
Si |
0.097 |
|
|
|
| 3 |
H |
0.013 |
|
|
|
| 4 |
H |
0.013 |
|
|
|
| 5 |
H |
-0.109 |
|
|
|
| 6 |
H |
-0.109 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
0.275 |
0.275 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-27.704 |
0.000 |
0.000 |
| y |
0.000 |
-31.864 |
0.000 |
| z |
0.000 |
0.000 |
-33.786 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
5.121 |
0.000 |
0.000 |
| y |
0.000 |
-1.119 |
0.000 |
| z |
0.000 |
0.000 |
-4.002 |
|
| Polar |
|---|
| 3z2-r2 | -8.004 |
|---|
| x2-y2 | 4.160 |
|---|
| xy | 0.000 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
8.366 |
0.000 |
0.000 |
| y |
0.000 |
15.152 |
0.000 |
| z |
0.000 |
0.000 |
10.115 |
<r2> (average value of r
2) Å
2
| <r2> |
80.004 |
| (<r2>)1/2 |
8.944 |