Jump to
S1C2
Energy calculated at BLYP/3-21G
| | hartrees |
|---|
| Energy at 0K | -321.754803 |
| Energy at 298.15K | |
| HF Energy | -321.754803 |
| Nuclear repulsion energy | 224.990825 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at BLYP/3-21G
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3042 |
3025 |
30.63 |
|
|
|
| 2 |
A' |
2993 |
2976 |
24.08 |
|
|
|
| 3 |
A' |
2974 |
2958 |
25.29 |
|
|
|
| 4 |
A' |
2965 |
2948 |
16.18 |
|
|
|
| 5 |
A' |
1539 |
1531 |
7.38 |
|
|
|
| 6 |
A' |
1527 |
1518 |
0.01 |
|
|
|
| 7 |
A' |
1517 |
1509 |
3.32 |
|
|
|
| 8 |
A' |
1429 |
1421 |
2.94 |
|
|
|
| 9 |
A' |
1389 |
1382 |
63.98 |
|
|
|
| 10 |
A' |
1349 |
1342 |
52.83 |
|
|
|
| 11 |
A' |
1304 |
1297 |
78.74 |
|
|
|
| 12 |
A' |
1119 |
1113 |
0.08 |
|
|
|
| 13 |
A' |
989 |
983 |
0.58 |
|
|
|
| 14 |
A' |
932 |
927 |
11.92 |
|
|
|
| 15 |
A' |
893 |
888 |
56.22 |
|
|
|
| 16 |
A' |
779 |
775 |
261.74 |
|
|
|
| 17 |
A' |
546 |
543 |
13.37 |
|
|
|
| 18 |
A' |
330 |
329 |
0.14 |
|
|
|
| 19 |
A' |
328 |
326 |
1.12 |
|
|
|
| 20 |
A' |
137 |
136 |
0.07 |
|
|
|
| 21 |
A" |
3047 |
3030 |
69.17 |
|
|
|
| 22 |
A" |
3019 |
3003 |
0.25 |
|
|
|
| 23 |
A" |
3001 |
2984 |
13.94 |
|
|
|
| 24 |
A" |
1534 |
1526 |
8.45 |
|
|
|
| 25 |
A" |
1322 |
1315 |
0.11 |
|
|
|
| 26 |
A" |
1247 |
1240 |
0.00 |
|
|
|
| 27 |
A" |
1134 |
1127 |
0.00 |
|
|
|
| 28 |
A" |
904 |
899 |
1.47 |
|
|
|
| 29 |
A" |
768 |
764 |
3.99 |
|
|
|
| 30 |
A" |
232 |
231 |
0.00 |
|
|
|
| 31 |
A" |
195 |
194 |
1.13 |
|
|
|
| 32 |
A" |
96 |
95 |
1.14 |
|
|
|
| 33 |
A" |
78i |
77i |
0.29 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 22250.2 cm
-1
Scaled (by 0.9945) Zero Point Vibrational Energy (zpe) 22127.8 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at BLYP/3-21G
Point Group is Cs
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
-1.854 |
2.374 |
0.000 |
| C2 |
-1.525 |
0.854 |
0.000 |
| C3 |
0.000 |
0.611 |
0.000 |
| O4 |
0.177 |
-0.880 |
0.000 |
| N5 |
1.735 |
-1.157 |
0.000 |
| O6 |
1.938 |
-2.374 |
0.000 |
| H7 |
-2.944 |
2.532 |
0.000 |
| H8 |
-1.435 |
2.866 |
0.894 |
| H9 |
-1.435 |
2.866 |
-0.894 |
| H10 |
-1.957 |
0.368 |
-0.890 |
| H11 |
-1.957 |
0.368 |
0.890 |
| H12 |
0.466 |
1.048 |
0.899 |
| H13 |
0.466 |
1.048 |
-0.899 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
C3 |
O4 |
N5 |
O6 |
H7 |
H8 |
H9 |
H10 |
H11 |
H12 |
H13 |
| C1 | | 1.5549 | 2.5585 | 3.8360 | 5.0344 | 6.0758 | 1.1018 | 1.1033 | 1.1033 | 2.1967 | 2.1967 | 2.8194 | 2.8194 |
C2 | 1.5549 | | 1.5439 | 2.4300 | 3.8300 | 4.7337 | 2.1977 | 2.2031 | 2.2031 | 1.1026 | 1.1026 | 2.1929 | 2.1929 | C3 | 2.5585 | 1.5439 | | 1.5013 | 2.4766 | 3.5581 | 3.5158 | 2.8186 | 2.8186 | 2.1638 | 2.1638 | 1.1030 | 1.1030 | O4 | 3.8360 | 2.4300 | 1.5013 | | 1.5821 | 2.3086 | 4.6246 | 4.1751 | 4.1751 | 2.6282 | 2.6282 | 2.1470 | 2.1470 | N5 | 5.0344 | 3.8300 | 2.4766 | 1.5821 | | 1.2337 | 5.9584 | 5.1988 | 5.1988 | 4.0928 | 4.0928 | 2.6978 | 2.6978 | O6 | 6.0758 | 4.7337 | 3.5581 | 2.3086 | 1.2337 | | 6.9210 | 6.2949 | 6.2949 | 4.8459 | 4.8459 | 3.8315 | 3.8315 | H7 | 1.1018 | 2.1977 | 3.5158 | 4.6246 | 5.9584 | 6.9210 | | 1.7860 | 1.7860 | 2.5393 | 2.5393 | 3.8265 | 3.8265 | H8 | 1.1033 | 2.2031 | 2.8186 | 4.1751 | 5.1988 | 6.2949 | 1.7860 | | 1.7887 | 3.1136 | 2.5516 | 2.6303 | 3.1834 | H9 | 1.1033 | 2.2031 | 2.8186 | 4.1751 | 5.1988 | 6.2949 | 1.7860 | 1.7887 | | 2.5516 | 3.1136 | 3.1834 | 2.6303 | H10 | 2.1967 | 1.1026 | 2.1638 | 2.6282 | 4.0928 | 4.8459 | 2.5393 | 3.1136 | 2.5516 | | 1.7802 | 3.0880 | 2.5171 | H11 | 2.1967 | 1.1026 | 2.1638 | 2.6282 | 4.0928 | 4.8459 | 2.5393 | 2.5516 | 3.1136 | 1.7802 | | 2.5171 | 3.0880 | H12 | 2.8194 | 2.1929 | 1.1030 | 2.1470 | 2.6978 | 3.8315 | 3.8265 | 2.6303 | 3.1834 | 3.0880 | 2.5171 | | 1.7977 | H13 | 2.8194 | 2.1929 | 1.1030 | 2.1470 | 2.6978 | 3.8315 | 3.8265 | 3.1834 | 2.6303 | 2.5171 | 3.0880 | 1.7977 | |
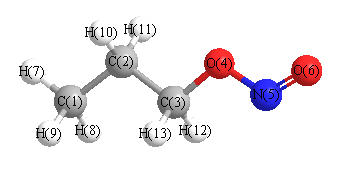 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
C3 |
111.310 |
|
C1 |
C2 |
H10 |
110.347 |
| C1 |
C2 |
H11 |
110.347 |
|
C2 |
C1 |
H7 |
110.474 |
| C2 |
C1 |
H8 |
110.810 |
|
C2 |
C1 |
H9 |
110.810 |
| C2 |
C3 |
O4 |
105.865 |
|
C2 |
C3 |
H12 |
110.781 |
| C2 |
C3 |
H13 |
110.781 |
|
C3 |
C2 |
H10 |
108.538 |
| C3 |
C2 |
H11 |
108.538 |
|
C3 |
O4 |
N5 |
106.845 |
| O4 |
C3 |
H12 |
110.115 |
|
O4 |
C3 |
H13 |
110.115 |
| O4 |
N5 |
O6 |
109.530 |
|
H7 |
C1 |
H8 |
108.173 |
| H7 |
C1 |
H9 |
108.173 |
|
H8 |
C1 |
H9 |
108.305 |
| H10 |
C2 |
H11 |
107.656 |
|
H12 |
C3 |
H13 |
109.149 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at BLYP/3-21G
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.527 |
|
|
|
| 2 |
C |
-0.377 |
|
|
|
| 3 |
C |
-0.129 |
|
|
|
| 4 |
O |
-0.306 |
|
|
|
| 5 |
N |
0.248 |
|
|
|
| 6 |
O |
-0.220 |
|
|
|
| 7 |
H |
0.188 |
|
|
|
| 8 |
H |
0.179 |
|
|
|
| 9 |
H |
0.179 |
|
|
|
| 10 |
H |
0.194 |
|
|
|
| 11 |
H |
0.194 |
|
|
|
| 12 |
H |
0.188 |
|
|
|
| 13 |
H |
0.188 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
-0.848 |
2.257 |
0.000 |
2.411 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-36.537 |
0.813 |
0.000 |
| y |
0.813 |
-38.747 |
0.000 |
| z |
0.000 |
0.000 |
-34.716 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
0.194 |
0.813 |
0.000 |
| y |
0.813 |
-3.121 |
0.000 |
| z |
0.000 |
0.000 |
2.926 |
|
| Polar |
|---|
| 3z2-r2 | 5.853 |
|---|
| x2-y2 | 2.210 |
|---|
| xy | 0.813 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
8.046 |
-2.816 |
0.000 |
| y |
-2.816 |
8.656 |
0.000 |
| z |
0.000 |
0.000 |
4.947 |
<r2> (average value of r
2) Å
2
| <r2> |
260.884 |
| (<r2>)1/2 |
16.152 |
Jump to
S1C1
Energy calculated at BLYP/3-21G
| | hartrees |
|---|
| Energy at 0K | -321.758884 |
| Energy at 298.15K | -321.767677 |
| HF Energy | -321.758884 |
| Nuclear repulsion energy | 233.176228 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at BLYP/3-21G
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3061 |
3045 |
25.03 |
|
|
|
| 2 |
A |
3041 |
3025 |
42.37 |
|
|
|
| 3 |
A |
3018 |
3002 |
23.86 |
|
|
|
| 4 |
A |
3006 |
2989 |
12.89 |
|
|
|
| 5 |
A |
2979 |
2963 |
15.56 |
|
|
|
| 6 |
A |
2976 |
2960 |
28.19 |
|
|
|
| 7 |
A |
2961 |
2945 |
22.62 |
|
|
|
| 8 |
A |
1544 |
1535 |
8.04 |
|
|
|
| 9 |
A |
1530 |
1522 |
7.78 |
|
|
|
| 10 |
A |
1506 |
1497 |
3.49 |
|
|
|
| 11 |
A |
1499 |
1491 |
1.93 |
|
|
|
| 12 |
A |
1419 |
1411 |
13.39 |
|
|
|
| 13 |
A |
1397 |
1389 |
81.23 |
|
|
|
| 14 |
A |
1366 |
1358 |
24.83 |
|
|
|
| 15 |
A |
1345 |
1338 |
72.57 |
|
|
|
| 16 |
A |
1291 |
1284 |
1.49 |
|
|
|
| 17 |
A |
1251 |
1244 |
3.26 |
|
|
|
| 18 |
A |
1150 |
1144 |
1.96 |
|
|
|
| 19 |
A |
1088 |
1082 |
4.87 |
|
|
|
| 20 |
A |
1008 |
1002 |
2.78 |
|
|
|
| 21 |
A |
928 |
923 |
17.56 |
|
|
|
| 22 |
A |
903 |
898 |
24.75 |
|
|
|
| 23 |
A |
835 |
830 |
4.17 |
|
|
|
| 24 |
A |
790 |
786 |
124.66 |
|
|
|
| 25 |
A |
757 |
753 |
109.15 |
|
|
|
| 26 |
A |
526 |
523 |
5.94 |
|
|
|
| 27 |
A |
420 |
417 |
6.29 |
|
|
|
| 28 |
A |
338 |
336 |
2.04 |
|
|
|
| 29 |
A |
262 |
261 |
1.78 |
|
|
|
| 30 |
A |
255 |
254 |
2.67 |
|
|
|
| 31 |
A |
181 |
180 |
0.36 |
|
|
|
| 32 |
A |
110 |
110 |
1.26 |
|
|
|
| 33 |
A |
69 |
69 |
0.13 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 22404.2 cm
-1
Scaled (by 0.9945) Zero Point Vibrational Energy (zpe) 22281.0 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at BLYP/3-21G
Point Group is C1
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
-2.215 |
-0.957 |
0.190 |
| C2 |
-1.828 |
0.454 |
-0.316 |
| C3 |
-0.465 |
0.919 |
0.224 |
| O4 |
0.549 |
-0.029 |
-0.305 |
| N5 |
1.859 |
0.223 |
0.320 |
| O6 |
2.703 |
-0.550 |
-0.141 |
| H7 |
-3.195 |
-1.264 |
-0.208 |
| H8 |
-2.271 |
-0.977 |
1.291 |
| H9 |
-1.461 |
-1.691 |
-0.131 |
| H10 |
-1.791 |
0.465 |
-1.418 |
| H11 |
-2.587 |
1.196 |
-0.007 |
| H12 |
-0.229 |
1.943 |
-0.111 |
| H13 |
-0.448 |
0.897 |
1.326 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
C3 |
O4 |
N5 |
O6 |
H7 |
H8 |
H9 |
H10 |
H11 |
H12 |
H13 |
| C1 | | 1.5480 | 2.5654 | 2.9570 | 4.2430 | 4.9452 | 1.1017 | 1.1033 | 1.1003 | 2.1879 | 2.1936 | 3.5273 | 2.8015 |
C2 | 1.5480 | | 1.5378 | 2.4260 | 3.7488 | 4.6441 | 2.1984 | 2.1975 | 2.1849 | 1.1029 | 1.1048 | 2.1944 | 2.1901 | C3 | 2.5654 | 1.5378 | | 1.4861 | 2.4285 | 3.5111 | 3.5221 | 2.8280 | 2.8168 | 2.1586 | 2.1519 | 1.1026 | 1.1029 | O4 | 2.9570 | 2.4260 | 1.4861 | | 1.4730 | 2.2218 | 3.9439 | 3.3765 | 2.6144 | 2.6380 | 3.3798 | 2.1284 | 2.1249 | N5 | 4.2430 | 3.7488 | 2.4285 | 1.4730 | | 1.2329 | 5.2946 | 4.4092 | 3.8588 | 4.0499 | 4.5629 | 2.7393 | 2.6066 | O6 | 4.9452 | 4.6441 | 3.5111 | 2.2218 | 1.2329 | | 5.9413 | 5.1933 | 4.3174 | 4.7807 | 5.5717 | 3.8480 | 3.7652 | H7 | 1.1017 | 2.1984 | 3.5221 | 3.9439 | 5.2946 | 5.9413 | | 1.7843 | 1.7877 | 2.5351 | 2.5419 | 4.3694 | 3.8168 | H8 | 1.1033 | 2.1975 | 2.8280 | 3.3765 | 4.4092 | 5.1933 | 1.7843 | | 1.7859 | 3.1069 | 2.5515 | 3.8296 | 2.6147 | H9 | 1.1003 | 2.1849 | 2.8168 | 2.6144 | 3.8588 | 4.3174 | 1.7877 | 1.7859 | | 2.5331 | 3.1016 | 3.8375 | 3.1387 | H10 | 2.1879 | 1.1029 | 2.1586 | 2.6380 | 4.0499 | 4.7807 | 2.5351 | 3.1069 | 2.5331 | | 1.7768 | 2.5161 | 3.0856 | H11 | 2.1936 | 1.1048 | 2.1519 | 3.3798 | 4.5629 | 5.5717 | 2.5419 | 2.5515 | 3.1016 | 1.7768 | | 2.4757 | 2.5378 | H12 | 3.5273 | 2.1944 | 1.1026 | 2.1284 | 2.7393 | 3.8480 | 4.3694 | 3.8296 | 3.8375 | 2.5161 | 2.4757 | | 1.7913 | H13 | 2.8015 | 2.1901 | 1.1029 | 2.1249 | 2.6066 | 3.7652 | 3.8168 | 2.6147 | 3.1387 | 3.0856 | 2.5378 | 1.7913 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
C3 |
112.474 |
|
C1 |
C2 |
H10 |
110.111 |
| C1 |
C2 |
H11 |
110.448 |
|
C2 |
C1 |
H7 |
111.007 |
| C2 |
C1 |
H8 |
110.848 |
|
C2 |
C1 |
H9 |
110.028 |
| C2 |
C3 |
O4 |
106.681 |
|
C2 |
C3 |
H12 |
111.353 |
| C2 |
C3 |
H13 |
110.994 |
|
C3 |
C2 |
H10 |
108.533 |
| C3 |
C2 |
H11 |
107.913 |
|
C3 |
O4 |
N5 |
110.312 |
| O4 |
C3 |
H12 |
109.724 |
|
O4 |
C3 |
H13 |
109.434 |
| O4 |
N5 |
O6 |
110.072 |
|
H7 |
C1 |
H8 |
108.038 |
| H7 |
C1 |
H9 |
108.554 |
|
H8 |
C1 |
H9 |
108.280 |
| H10 |
C2 |
H11 |
107.186 |
|
H12 |
C3 |
H13 |
108.628 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at BLYP/3-21G
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.519 |
|
|
|
| 2 |
C |
-0.372 |
|
|
|
| 3 |
C |
-0.142 |
|
|
|
| 4 |
O |
-0.294 |
|
|
|
| 5 |
N |
0.253 |
|
|
|
| 6 |
O |
-0.218 |
|
|
|
| 7 |
H |
0.179 |
|
|
|
| 8 |
H |
0.175 |
|
|
|
| 9 |
H |
0.187 |
|
|
|
| 10 |
H |
0.192 |
|
|
|
| 11 |
H |
0.179 |
|
|
|
| 12 |
H |
0.193 |
|
|
|
| 13 |
H |
0.188 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
1.782 |
-0.618 |
0.905 |
2.092 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-37.453 |
0.032 |
-0.795 |
| y |
0.032 |
-34.646 |
-0.745 |
| z |
-0.795 |
-0.745 |
-36.756 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-1.753 |
0.032 |
-0.795 |
| y |
0.032 |
2.458 |
-0.745 |
| z |
-0.795 |
-0.745 |
-0.706 |
|
| Polar |
|---|
| 3z2-r2 | -1.412 |
|---|
| x2-y2 | -2.807 |
|---|
| xy | 0.032 |
|---|
| xz | -0.795 |
|---|
| yz | -0.745 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
9.543 |
-1.433 |
-0.056 |
| y |
-1.433 |
6.214 |
0.001 |
| z |
-0.056 |
0.001 |
5.348 |
<r2> (average value of r
2) Å
2
| <r2> |
203.556 |
| (<r2>)1/2 |
14.267 |