Jump to
S1C2
S1C3
S1C4
Energy calculated at BLYP/6-31+G**
| | hartrees |
|---|
| Energy at 0K | -579.998283 |
| Energy at 298.15K | |
| HF Energy | -579.998283 |
| Nuclear repulsion energy | 66.436104 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at BLYP/6-31+G**
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
Σg |
2280 |
2268 |
0.00 |
347.44 |
0.33 |
0.49 |
| 2 |
Σg |
726 |
722 |
0.00 |
115.26 |
0.13 |
0.24 |
| 3 |
Σu |
2278 |
2266 |
0.05 |
0.00 |
0.00 |
0.00 |
| 4 |
Πg |
642i |
639i |
0.00 |
57.17 |
0.75 |
0.86 |
| 4 |
Πg |
642i |
639i |
0.00 |
57.17 |
0.75 |
0.86 |
| 5 |
Πu |
388 |
385 |
6.12 |
0.00 |
0.00 |
0.00 |
| 5 |
Πu |
388 |
385 |
6.12 |
0.00 |
0.00 |
0.00 |
Unscaled Zero Point Vibrational Energy (zpe) 2386.6 cm
-1
Scaled (by 0.9947) Zero Point Vibrational Energy (zpe) 2374.0 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at BLYP/6-31+G**
Point Group is D∞h
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| Si1 |
0.000 |
0.000 |
0.997 |
| Si2 |
0.000 |
0.000 |
-0.997 |
| H3 |
0.000 |
0.000 |
2.470 |
| H4 |
0.000 |
0.000 |
-2.470 |
Atom - Atom Distances (Å)
| |
Si1 |
Si2 |
H3 |
H4 |
| Si1 | | 1.9948 | 1.4725 | 3.4674 |
Si2 | 1.9948 | | 3.4674 | 1.4725 | H3 | 1.4725 | 3.4674 | | 4.9399 | H4 | 3.4674 | 1.4725 | 4.9399 | |
 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| Si1 |
Si2 |
H4 |
180.000 |
|
Si2 |
Si1 |
H3 |
180.000 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at BLYP/6-31+G**
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
Si |
-0.014 |
|
|
|
| 2 |
Si |
-0.014 |
|
|
|
| 3 |
H |
0.014 |
|
|
|
| 4 |
H |
0.014 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
0.000 |
0.000 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-30.536 |
0.000 |
0.000 |
| y |
0.000 |
-30.536 |
0.000 |
| z |
0.000 |
0.000 |
-20.914 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-4.811 |
0.000 |
0.000 |
| y |
0.000 |
-4.811 |
0.000 |
| z |
0.000 |
0.000 |
9.622 |
|
| Polar |
|---|
| 3z2-r2 | 19.244 |
|---|
| x2-y2 | 0.000 |
|---|
| xy | 0.000 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
6.523 |
0.000 |
0.000 |
| y |
0.000 |
6.523 |
0.000 |
| z |
0.000 |
0.000 |
15.671 |
<r2> (average value of r
2) Å
2
| <r2> |
57.126 |
| (<r2>)1/2 |
7.558 |
Jump to
S1C1
S1C3
S1C4
Energy calculated at BLYP/6-31+G**
| | hartrees |
|---|
| Energy at 0K | -580.036833 |
| Energy at 298.15K | |
| HF Energy | -580.036833 |
| Nuclear repulsion energy | 63.098816 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at BLYP/6-31+G**
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
Ag |
2096 |
2085 |
0.00 |
457.22 |
0.49 |
0.66 |
| 2 |
Ag |
619 |
616 |
0.00 |
43.59 |
0.61 |
0.76 |
| 3 |
Ag |
541 |
538 |
0.00 |
82.39 |
0.09 |
0.17 |
| 4 |
Au |
221 |
219 |
63.42 |
0.00 |
0.00 |
0.00 |
| 5 |
Bu |
2102 |
2091 |
173.66 |
0.00 |
0.00 |
0.00 |
| 6 |
Bu |
233 |
232 |
31.91 |
0.00 |
0.00 |
0.00 |
Unscaled Zero Point Vibrational Energy (zpe) 2905.9 cm
-1
Scaled (by 0.9947) Zero Point Vibrational Energy (zpe) 2890.5 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at BLYP/6-31+G**
Point Group is C2h
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| Si1 |
0.000 |
1.068 |
0.000 |
| Si2 |
0.000 |
-1.068 |
0.000 |
| H3 |
1.254 |
1.906 |
0.000 |
| H4 |
-1.254 |
-1.906 |
0.000 |
Atom - Atom Distances (Å)
| |
Si1 |
Si2 |
H3 |
H4 |
| Si1 | | 2.1355 | 1.5085 | 3.2274 |
Si2 | 2.1355 | | 3.2274 | 1.5085 | H3 | 1.5085 | 3.2274 | | 4.5632 | H4 | 3.2274 | 1.5085 | 4.5632 | |
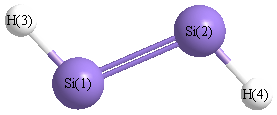 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| Si1 |
Si2 |
H4 |
123.764 |
|
Si2 |
Si1 |
H3 |
123.764 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at BLYP/6-31+G**
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
Si |
0.041 |
|
|
|
| 2 |
Si |
0.041 |
|
|
|
| 3 |
H |
-0.041 |
|
|
|
| 4 |
H |
-0.041 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
0.000 |
0.000 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-29.010 |
-0.244 |
0.000 |
| y |
-0.244 |
-23.929 |
0.000 |
| z |
0.000 |
0.000 |
-31.274 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-1.408 |
-0.244 |
0.000 |
| y |
-0.244 |
6.212 |
0.000 |
| z |
0.000 |
0.000 |
-4.804 |
|
| Polar |
|---|
| 3z2-r2 | -9.608 |
|---|
| x2-y2 | -5.081 |
|---|
| xy | -0.244 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
7.074 |
0.213 |
0.000 |
| y |
0.213 |
16.182 |
0.000 |
| z |
0.000 |
0.000 |
8.344 |
<r2> (average value of r
2) Å
2
| <r2> |
59.866 |
| (<r2>)1/2 |
7.737 |
Jump to
S1C1
S1C2
S1C4
Energy calculated at BLYP/6-31+G**
| | hartrees |
|---|
| Energy at 0K | -580.061334 |
| Energy at 298.15K | |
| HF Energy | -580.061334 |
| Nuclear repulsion energy | 63.805007 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at BLYP/6-31+G**
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A1 |
1564 |
1556 |
10.23 |
81.44 |
0.05 |
0.10 |
| 2 |
A1 |
924 |
919 |
41.64 |
1.72 |
0.15 |
0.26 |
| 3 |
A1 |
488 |
485 |
0.87 |
59.15 |
0.34 |
0.50 |
| 4 |
A2 |
1021 |
1016 |
0.00 |
7.12 |
0.75 |
0.86 |
| 5 |
B1 |
1481 |
1473 |
19.41 |
10.80 |
0.75 |
0.86 |
| 6 |
B2 |
1101 |
1095 |
345.41 |
1.42 |
0.75 |
0.86 |
Unscaled Zero Point Vibrational Energy (zpe) 3289.1 cm
-1
Scaled (by 0.9947) Zero Point Vibrational Energy (zpe) 3271.7 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at BLYP/6-31+G**
Point Group is C2v
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| Si1 |
0.000 |
1.125 |
-0.052 |
| Si2 |
0.000 |
-1.125 |
-0.052 |
| H3 |
0.999 |
0.000 |
0.735 |
| H4 |
-0.999 |
0.000 |
0.735 |
Atom - Atom Distances (Å)
| |
Si1 |
Si2 |
H3 |
H4 |
| Si1 | | 2.2505 | 1.6979 | 1.6979 |
Si2 | 2.2505 | | 1.6979 | 1.6979 | H3 | 1.6979 | 1.6979 | | 1.9971 | H4 | 1.6979 | 1.6979 | 1.9971 | |
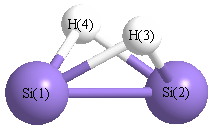 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| Si1 |
Si2 |
H4 |
48.493 |
|
Si2 |
Si1 |
H3 |
48.493 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at BLYP/6-31+G**
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
Si |
0.124 |
|
|
|
| 2 |
Si |
0.124 |
|
|
|
| 3 |
H |
-0.124 |
|
|
|
| 4 |
H |
-0.124 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
-0.549 |
0.549 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-26.448 |
0.000 |
0.000 |
| y |
0.000 |
-32.359 |
0.000 |
| z |
0.000 |
0.000 |
-29.363 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
4.413 |
0.000 |
0.000 |
| y |
0.000 |
-4.453 |
0.000 |
| z |
0.000 |
0.000 |
0.041 |
|
| Polar |
|---|
| 3z2-r2 | 0.081 |
|---|
| x2-y2 | 5.911 |
|---|
| xy | 0.000 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
6.657 |
0.000 |
0.000 |
| y |
0.000 |
15.147 |
0.000 |
| z |
0.000 |
0.000 |
7.325 |
<r2> (average value of r
2) Å
2
| <r2> |
56.961 |
| (<r2>)1/2 |
7.547 |
Jump to
S1C1
S1C2
S1C3
Energy calculated at BLYP/6-31+G**
| | hartrees |
|---|
| Energy at 0K | -580.047383 |
| Energy at 298.15K | |
| HF Energy | -580.047383 |
| Nuclear repulsion energy | 64.165957 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at BLYP/6-31+G**
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
2094 |
2083 |
116.45 |
344.02 |
0.42 |
0.59 |
| 2 |
A' |
1583 |
1575 |
91.67 |
91.45 |
0.12 |
0.22 |
| 3 |
A' |
1003 |
998 |
118.15 |
6.00 |
0.44 |
0.61 |
| 4 |
A' |
582 |
579 |
14.68 |
67.89 |
0.23 |
0.37 |
| 5 |
A' |
441 |
439 |
5.18 |
5.88 |
0.15 |
0.26 |
| 6 |
A" |
94 |
93 |
50.70 |
6.16 |
0.75 |
0.86 |
Unscaled Zero Point Vibrational Energy (zpe) 2898.8 cm
-1
Scaled (by 0.9947) Zero Point Vibrational Energy (zpe) 2883.4 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at BLYP/6-31+G**
Point Group is Cs
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| Si1 |
0.062 |
-1.159 |
0.000 |
| Si2 |
0.062 |
0.989 |
0.000 |
| H3 |
-1.258 |
-0.012 |
0.000 |
| H4 |
-0.474 |
2.397 |
0.000 |
Atom - Atom Distances (Å)
| |
Si1 |
Si2 |
H3 |
H4 |
| Si1 | | 2.1486 | 1.7491 | 3.5969 |
Si2 | 2.1486 | | 1.6570 | 1.5067 | H3 | 1.7491 | 1.6570 | | 2.5340 | H4 | 3.5969 | 1.5067 | 2.5340 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| Si1 |
Si2 |
H4 |
159.171 |
|
Si2 |
Si1 |
H3 |
49.009 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at BLYP/6-31+G**
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
Si |
0.124 |
|
|
|
| 2 |
Si |
0.014 |
|
|
|
| 3 |
H |
-0.102 |
|
|
|
| 4 |
H |
-0.036 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
-0.011 |
0.654 |
0.000 |
0.654 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-27.182 |
0.289 |
0.000 |
| y |
0.289 |
-26.336 |
0.000 |
| z |
0.000 |
0.000 |
-31.471 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
1.722 |
0.289 |
0.000 |
| y |
0.289 |
2.990 |
0.000 |
| z |
0.000 |
0.000 |
-4.712 |
|
| Polar |
|---|
| 3z2-r2 | -9.424 |
|---|
| x2-y2 | -0.845 |
|---|
| xy | 0.289 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
6.282 |
-0.083 |
0.000 |
| y |
-0.083 |
15.632 |
0.000 |
| z |
0.000 |
0.000 |
8.084 |
<r2> (average value of r
2) Å
2
| <r2> |
57.874 |
| (<r2>)1/2 |
7.607 |