Jump to
S1C2
Energy calculated at BLYP/6-31+G**
| | hartrees |
|---|
| Energy at 0K | -358.425809 |
| Energy at 298.15K | -358.430657 |
| Nuclear repulsion energy | 229.055505 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at BLYP/6-31+G**
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3626 |
3607 |
71.47 |
|
|
|
| 2 |
A' |
3593 |
3574 |
58.24 |
|
|
|
| 3 |
A' |
3483 |
3464 |
45.76 |
|
|
|
| 4 |
A' |
1716 |
1707 |
108.73 |
|
|
|
| 5 |
A' |
1703 |
1694 |
474.16 |
|
|
|
| 6 |
A' |
1550 |
1542 |
108.58 |
|
|
|
| 7 |
A' |
1343 |
1336 |
4.93 |
|
|
|
| 8 |
A' |
1262 |
1255 |
56.56 |
|
|
|
| 9 |
A' |
1123 |
1117 |
281.12 |
|
|
|
| 10 |
A' |
1051 |
1046 |
4.37 |
|
|
|
| 11 |
A' |
735 |
731 |
5.51 |
|
|
|
| 12 |
A' |
586 |
582 |
64.35 |
|
|
|
| 13 |
A' |
501 |
498 |
0.58 |
|
|
|
| 14 |
A' |
397 |
394 |
4.41 |
|
|
|
| 15 |
A' |
258 |
257 |
14.31 |
|
|
|
| 16 |
A" |
785 |
780 |
6.17 |
|
|
|
| 17 |
A" |
655 |
652 |
122.30 |
|
|
|
| 18 |
A" |
613 |
610 |
12.80 |
|
|
|
| 19 |
A" |
415 |
413 |
9.99 |
|
|
|
| 20 |
A" |
330 |
329 |
207.48 |
|
|
|
| 21 |
A" |
63 |
63 |
4.72 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 12893.5 cm
-1
Scaled (by 0.9947) Zero Point Vibrational Energy (zpe) 12825.2 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at BLYP/6-31+G**
Point Group is Cs
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.000 |
0.763 |
0.000 |
| C2 |
-0.052 |
-0.796 |
0.000 |
| O3 |
-1.105 |
-1.435 |
0.000 |
| O4 |
1.046 |
1.408 |
0.000 |
| O5 |
-1.239 |
1.312 |
0.000 |
| N6 |
1.209 |
-1.329 |
0.000 |
| H7 |
1.318 |
-2.340 |
0.000 |
| H8 |
2.025 |
-0.720 |
0.000 |
| H9 |
-1.115 |
2.288 |
0.000 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
O3 |
O4 |
O5 |
N6 |
H7 |
H8 |
H9 |
| C1 | | 1.5604 | 2.4608 | 1.2290 | 1.3545 | 2.4167 | 3.3717 | 2.5099 | 1.8890 |
C2 | 1.5604 | | 1.2323 | 2.4624 | 2.4190 | 1.3685 | 2.0638 | 2.0776 | 3.2625 | O3 | 2.4608 | 1.2323 | | 3.5655 | 2.7501 | 2.3164 | 2.5866 | 3.2105 | 3.7234 | O4 | 1.2290 | 2.4624 | 3.5655 | | 2.2871 | 2.7419 | 3.7577 | 2.3419 | 2.3339 | O5 | 1.3545 | 2.4190 | 2.7501 | 2.2871 | | 3.6005 | 4.4576 | 3.8439 | 0.9843 | N6 | 2.4167 | 1.3685 | 2.3164 | 2.7419 | 3.6005 | | 1.0167 | 1.0182 | 4.2995 | H7 | 3.3717 | 2.0638 | 2.5866 | 3.7577 | 4.4576 | 1.0167 | | 1.7674 | 5.2287 | H8 | 2.5099 | 2.0776 | 3.2105 | 2.3419 | 3.8439 | 1.0182 | 1.7674 | | 4.3482 | H9 | 1.8890 | 3.2625 | 3.7234 | 2.3339 | 0.9843 | 4.2995 | 5.2287 | 4.3482 | |
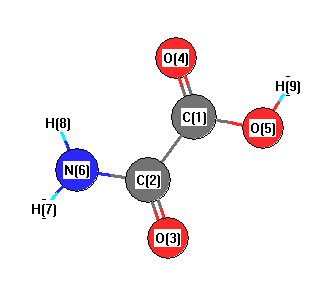 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
O3 |
123.137 |
|
C1 |
C2 |
N6 |
111.030 |
| C1 |
O5 |
H9 |
106.665 |
|
C2 |
C1 |
O4 |
123.522 |
| C2 |
C1 |
O5 |
111.978 |
|
C2 |
N6 |
H7 |
119.093 |
| C2 |
N6 |
H8 |
120.323 |
|
O3 |
C2 |
N6 |
125.832 |
| O4 |
C1 |
O5 |
124.500 |
|
H7 |
N6 |
H8 |
120.585 |
Electronic energy levels
Electronic state
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at BLYP/6-31+G**
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
0.392 |
|
|
|
| 2 |
C |
0.454 |
|
|
|
| 3 |
O |
-0.499 |
|
|
|
| 4 |
O |
-0.475 |
|
|
|
| 5 |
O |
-0.371 |
|
|
|
| 6 |
N |
-0.497 |
|
|
|
| 7 |
H |
0.314 |
|
|
|
| 8 |
H |
0.318 |
|
|
|
| 9 |
H |
0.362 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
2.277 |
0.600 |
0.000 |
2.355 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-25.557 |
8.383 |
0.003 |
| y |
8.383 |
-40.294 |
0.005 |
| z |
0.003 |
0.005 |
-34.615 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
11.897 |
8.383 |
0.003 |
| y |
8.383 |
-10.207 |
0.005 |
| z |
0.003 |
0.005 |
-1.690 |
|
| Polar |
|---|
| 3z2-r2 | -3.380 |
|---|
| x2-y2 | 14.736 |
|---|
| xy | 8.383 |
|---|
| xz | 0.003 |
|---|
| yz | 0.005 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
0.000 |
0.000 |
0.000 |
| y |
0.000 |
0.000 |
0.000 |
| z |
0.000 |
0.000 |
0.000 |
<r2> (average value of r
2) Å
2
| <r2> |
146.023 |
| (<r2>)1/2 |
12.084 |
Jump to
S1C1
Energy calculated at BLYP/6-31+G**
| | hartrees |
|---|
| Energy at 0K | -358.431528 |
| Energy at 298.15K | -358.436598 |
| Nuclear repulsion energy | 230.448518 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at BLYP/6-31+G**
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3617 |
3598 |
76.28 |
|
|
|
| 2 |
A' |
3477 |
3459 |
57.10 |
|
|
|
| 3 |
A' |
3416 |
3398 |
126.97 |
|
|
|
| 4 |
A' |
1759 |
1749 |
247.86 |
|
|
|
| 5 |
A' |
1692 |
1684 |
302.64 |
|
|
|
| 6 |
A' |
1553 |
1544 |
64.21 |
|
|
|
| 7 |
A' |
1363 |
1356 |
146.83 |
|
|
|
| 8 |
A' |
1286 |
1280 |
354.05 |
|
|
|
| 9 |
A' |
1144 |
1138 |
20.44 |
|
|
|
| 10 |
A' |
1058 |
1052 |
2.93 |
|
|
|
| 11 |
A' |
756 |
752 |
10.04 |
|
|
|
| 12 |
A' |
602 |
599 |
10.82 |
|
|
|
| 13 |
A' |
516 |
514 |
1.88 |
|
|
|
| 14 |
A' |
384 |
382 |
6.01 |
|
|
|
| 15 |
A' |
255 |
254 |
41.51 |
|
|
|
| 16 |
A" |
776 |
772 |
4.84 |
|
|
|
| 17 |
A" |
727 |
724 |
98.07 |
|
|
|
| 18 |
A" |
637 |
633 |
3.50 |
|
|
|
| 19 |
A" |
441 |
439 |
116.82 |
|
|
|
| 20 |
A" |
377 |
375 |
119.39 |
|
|
|
| 21 |
A" |
110 |
110 |
7.61 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 12972.9 cm
-1
Scaled (by 0.9947) Zero Point Vibrational Energy (zpe) 12904.2 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at BLYP/6-31+G**
Point Group is Cs
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.015 |
-0.805 |
0.000 |
| C2 |
0.000 |
0.756 |
0.000 |
| O3 |
-1.088 |
1.359 |
0.000 |
| O4 |
1.038 |
-1.475 |
0.000 |
| O5 |
-1.235 |
-1.315 |
0.000 |
| N6 |
1.234 |
1.317 |
0.000 |
| H7 |
1.335 |
2.329 |
0.000 |
| H8 |
2.056 |
0.714 |
0.000 |
| H9 |
-1.846 |
-0.530 |
0.000 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
O3 |
O4 |
O5 |
N6 |
H7 |
H8 |
H9 |
| C1 | | 1.5609 | 2.4288 | 1.2227 | 1.3503 | 2.4465 | 3.4001 | 2.5438 | 1.8811 |
C2 | 1.5609 | | 1.2438 | 2.4605 | 2.4111 | 1.3555 | 2.0629 | 2.0565 | 2.2498 | O3 | 2.4288 | 1.2438 | | 3.5426 | 2.6779 | 2.3226 | 2.6098 | 3.2094 | 2.0358 | O4 | 1.2227 | 2.4605 | 3.5426 | | 2.2786 | 2.7981 | 3.8150 | 2.4138 | 3.0344 | O5 | 1.3503 | 2.4111 | 2.6779 | 2.2786 | | 3.6084 | 4.4587 | 3.8660 | 0.9941 | N6 | 2.4465 | 1.3555 | 2.3226 | 2.7981 | 3.6084 | | 1.0173 | 1.0190 | 3.5913 | H7 | 3.4001 | 2.0629 | 2.6098 | 3.8150 | 4.4587 | 1.0173 | | 1.7684 | 4.2769 | H8 | 2.5438 | 2.0565 | 3.2094 | 2.4138 | 3.8660 | 1.0190 | 1.7684 | | 4.0954 | H9 | 1.8811 | 2.2498 | 2.0358 | 3.0344 | 0.9941 | 3.5913 | 4.2769 | 4.0954 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
O3 |
119.561 |
|
C1 |
C2 |
N6 |
113.855 |
| C1 |
O5 |
H9 |
105.718 |
|
C2 |
C1 |
O4 |
123.790 |
| C2 |
C1 |
O5 |
111.628 |
|
C2 |
N6 |
H7 |
120.104 |
| C2 |
N6 |
H8 |
119.333 |
|
O3 |
C2 |
N6 |
126.584 |
| O4 |
C1 |
O5 |
124.581 |
|
H7 |
N6 |
H8 |
120.564 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at BLYP/6-31+G**
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
0.467 |
|
|
|
| 2 |
C |
0.416 |
|
|
|
| 3 |
O |
-0.548 |
|
|
|
| 4 |
O |
-0.472 |
|
|
|
| 5 |
O |
-0.381 |
|
|
|
| 6 |
N |
-0.490 |
|
|
|
| 7 |
H |
0.318 |
|
|
|
| 8 |
H |
0.325 |
|
|
|
| 9 |
H |
0.365 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
1.607 |
2.716 |
0.000 |
3.155 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-31.372 |
7.672 |
0.000 |
| y |
7.672 |
-38.729 |
0.000 |
| z |
0.000 |
0.000 |
-34.529 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
5.257 |
7.672 |
0.000 |
| y |
7.672 |
-5.778 |
0.000 |
| z |
0.000 |
0.000 |
0.521 |
|
| Polar |
|---|
| 3z2-r2 | 1.042 |
|---|
| x2-y2 | 7.357 |
|---|
| xy | 7.672 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
8.550 |
-0.001 |
0.000 |
| y |
-0.001 |
7.157 |
0.000 |
| z |
0.000 |
0.000 |
4.037 |
<r2> (average value of r
2) Å
2
| <r2> |
143.819 |
| (<r2>)1/2 |
11.992 |