Vibrational Frequencies calculated at BLYP/cc-pVDZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3623 |
3629 |
7.90 |
|
|
|
| 2 |
A |
3613 |
3618 |
6.15 |
|
|
|
| 3 |
A |
3027 |
3032 |
35.87 |
|
|
|
| 4 |
A |
3006 |
3011 |
40.66 |
|
|
|
| 5 |
A |
2998 |
3002 |
30.42 |
|
|
|
| 6 |
A |
2955 |
2960 |
27.65 |
|
|
|
| 7 |
A |
2943 |
2947 |
22.08 |
|
|
|
| 8 |
A |
2932 |
2936 |
35.60 |
|
|
|
| 9 |
A |
2855 |
2860 |
133.85 |
|
|
|
| 10 |
A |
2845 |
2849 |
13.08 |
|
|
|
| 11 |
A |
1461 |
1463 |
2.77 |
|
|
|
| 12 |
A |
1432 |
1434 |
4.76 |
|
|
|
| 13 |
A |
1424 |
1426 |
3.76 |
|
|
|
| 14 |
A |
1417 |
1419 |
1.14 |
|
|
|
| 15 |
A |
1402 |
1405 |
8.43 |
|
|
|
| 16 |
A |
1372 |
1374 |
17.99 |
|
|
|
| 17 |
A |
1350 |
1352 |
5.93 |
|
|
|
| 18 |
A |
1321 |
1323 |
6.33 |
|
|
|
| 19 |
A |
1282 |
1284 |
25.39 |
|
|
|
| 20 |
A |
1271 |
1273 |
4.55 |
|
|
|
| 21 |
A |
1248 |
1250 |
29.32 |
|
|
|
| 22 |
A |
1210 |
1212 |
45.46 |
|
|
|
| 23 |
A |
1162 |
1164 |
3.09 |
|
|
|
| 24 |
A |
1106 |
1108 |
7.60 |
|
|
|
| 25 |
A |
1081 |
1083 |
49.96 |
|
|
|
| 26 |
A |
1049 |
1051 |
1.93 |
|
|
|
| 27 |
A |
1009 |
1010 |
90.53 |
|
|
|
| 28 |
A |
986 |
987 |
14.03 |
|
|
|
| 29 |
A |
950 |
951 |
9.26 |
|
|
|
| 30 |
A |
907 |
909 |
10.53 |
|
|
|
| 31 |
A |
826 |
827 |
16.08 |
|
|
|
| 32 |
A |
783 |
785 |
7.07 |
|
|
|
| 33 |
A |
489 |
489 |
15.12 |
|
|
|
| 34 |
A |
463 |
463 |
6.58 |
|
|
|
| 35 |
A |
382 |
382 |
5.95 |
|
|
|
| 36 |
A |
329 |
329 |
7.16 |
|
|
|
| 37 |
A |
296 |
296 |
97.23 |
|
|
|
| 38 |
A |
261 |
262 |
82.11 |
|
|
|
| 39 |
A |
245 |
246 |
0.54 |
|
|
|
| 40 |
A |
176 |
176 |
3.04 |
|
|
|
| 41 |
A |
114 |
115 |
5.63 |
|
|
|
| 42 |
A |
93 |
93 |
6.03 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 29845.0 cm
-1
Scaled (by 1.0016) Zero Point Vibrational Energy (zpe) 29892.7 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
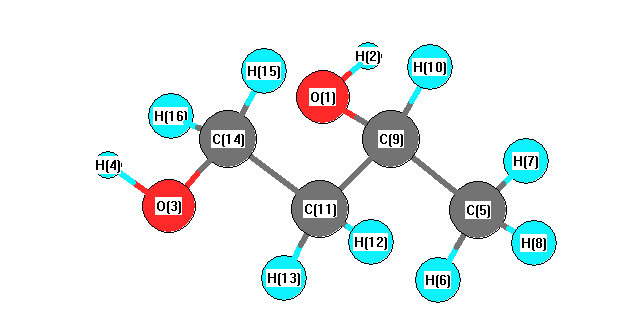
Charges from optimized geometry at BLYP/cc-pVDZ
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
O |
-0.236 |
|
|
|
| 2 |
H |
0.126 |
|
|
|
| 3 |
O |
-0.243 |
|
|
|
| 4 |
H |
0.128 |
|
|
|
| 5 |
C |
0.058 |
|
|
|
| 6 |
H |
0.011 |
|
|
|
| 7 |
H |
-0.012 |
|
|
|
| 8 |
H |
0.002 |
|
|
|
| 9 |
C |
0.032 |
|
|
|
| 10 |
H |
-0.031 |
|
|
|
| 11 |
C |
0.064 |
|
|
|
| 12 |
H |
-0.009 |
|
|
|
| 13 |
H |
-0.003 |
|
|
|
| 14 |
C |
0.134 |
|
|
|
| 15 |
H |
-0.022 |
|
|
|
| 16 |
H |
-0.001 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.960 |
0.817 |
1.277 |
1.795 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-32.059 |
-2.225 |
-0.268 |
| y |
-2.225 |
-38.644 |
0.802 |
| z |
-0.268 |
0.802 |
-39.038 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
6.782 |
-2.225 |
-0.268 |
| y |
-2.225 |
-3.096 |
0.802 |
| z |
-0.268 |
0.802 |
-3.686 |
|
| Polar |
|---|
| 3z2-r2 | -7.372 |
|---|
| x2-y2 | 6.585 |
|---|
| xy | -2.225 |
|---|
| xz | -0.268 |
|---|
| yz | 0.802 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
9.949 |
-0.201 |
0.074 |
| y |
-0.201 |
8.000 |
0.115 |
| z |
0.074 |
0.115 |
7.237 |
<r2> (average value of r
2) Å
2
| <r2> |
0.000 |
| (<r2>)1/2 |
0.000 |