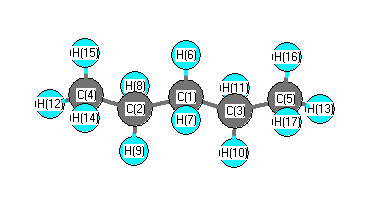
Vibrational Frequencies calculated at BLYP/cc-pVDZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A1 |
3013 |
3018 |
53.06 |
|
|
|
| 2 |
A1 |
2945 |
2949 |
29.47 |
|
|
|
| 3 |
A1 |
2932 |
2937 |
91.29 |
|
|
|
| 4 |
A1 |
2915 |
2920 |
8.47 |
|
|
|
| 5 |
A1 |
1449 |
1452 |
2.36 |
|
|
|
| 6 |
A1 |
1431 |
1433 |
0.69 |
|
|
|
| 7 |
A1 |
1420 |
1422 |
0.02 |
|
|
|
| 8 |
A1 |
1361 |
1363 |
0.65 |
|
|
|
| 9 |
A1 |
1321 |
1323 |
0.22 |
|
|
|
| 10 |
A1 |
1122 |
1124 |
0.92 |
|
|
|
| 11 |
A1 |
1016 |
1018 |
0.50 |
|
|
|
| 12 |
A1 |
848 |
849 |
0.88 |
|
|
|
| 13 |
A1 |
387 |
388 |
0.00 |
|
|
|
| 14 |
A1 |
172 |
173 |
0.02 |
|
|
|
| 15 |
A2 |
3007 |
3012 |
0.00 |
|
|
|
| 16 |
A2 |
2952 |
2957 |
0.00 |
|
|
|
| 17 |
A2 |
1431 |
1434 |
0.00 |
|
|
|
| 18 |
A2 |
1288 |
1290 |
0.00 |
|
|
|
| 19 |
A2 |
1222 |
1224 |
0.00 |
|
|
|
| 20 |
A2 |
964 |
966 |
0.00 |
|
|
|
| 21 |
A2 |
751 |
752 |
0.00 |
|
|
|
| 22 |
A2 |
250 |
251 |
0.00 |
|
|
|
| 23 |
A2 |
115 |
115 |
0.00 |
|
|
|
| 24 |
B1 |
3008 |
3013 |
126.34 |
|
|
|
| 25 |
B1 |
2969 |
2974 |
59.68 |
|
|
|
| 26 |
B1 |
2934 |
2939 |
0.28 |
|
|
|
| 27 |
B1 |
1430 |
1432 |
9.54 |
|
|
|
| 28 |
B1 |
1283 |
1285 |
0.45 |
|
|
|
| 29 |
B1 |
1163 |
1165 |
0.12 |
|
|
|
| 30 |
B1 |
844 |
846 |
1.26 |
|
|
|
| 31 |
B1 |
726 |
727 |
3.59 |
|
|
|
| 32 |
B1 |
243 |
243 |
0.00 |
|
|
|
| 33 |
B1 |
117 |
117 |
0.00 |
|
|
|
| 34 |
B2 |
3013 |
3018 |
34.67 |
|
|
|
| 35 |
B2 |
2944 |
2948 |
48.28 |
|
|
|
| 36 |
B2 |
2925 |
2930 |
0.18 |
|
|
|
| 37 |
B2 |
1441 |
1443 |
0.60 |
|
|
|
| 38 |
B2 |
1421 |
1423 |
0.69 |
|
|
|
| 39 |
B2 |
1359 |
1361 |
0.36 |
|
|
|
| 40 |
B2 |
1348 |
1350 |
2.10 |
|
|
|
| 41 |
B2 |
1249 |
1251 |
10.58 |
|
|
|
| 42 |
B2 |
1044 |
1046 |
0.63 |
|
|
|
| 43 |
B2 |
1005 |
1007 |
0.45 |
|
|
|
| 44 |
B2 |
901 |
903 |
1.80 |
|
|
|
| 45 |
B2 |
385 |
386 |
0.01 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 34030.9 cm
-1
Scaled (by 1.0016) Zero Point Vibrational Energy (zpe) 34085.4 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at BLYP/cc-pVDZ
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
0.019 |
|
|
|
| 2 |
C |
-0.004 |
|
|
|
| 3 |
C |
-0.004 |
|
|
|
| 4 |
C |
0.024 |
|
|
|
| 5 |
C |
0.024 |
|
|
|
| 6 |
H |
-0.014 |
|
|
|
| 7 |
H |
-0.014 |
|
|
|
| 8 |
H |
-0.011 |
|
|
|
| 9 |
H |
-0.011 |
|
|
|
| 10 |
H |
-0.011 |
|
|
|
| 11 |
H |
-0.011 |
|
|
|
| 12 |
H |
-0.002 |
|
|
|
| 13 |
H |
-0.002 |
|
|
|
| 14 |
H |
0.004 |
|
|
|
| 15 |
H |
0.004 |
|
|
|
| 16 |
H |
0.004 |
|
|
|
| 17 |
H |
0.004 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
-0.066 |
0.066 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-34.971 |
0.000 |
0.000 |
| y |
0.000 |
-36.143 |
0.000 |
| z |
0.000 |
0.000 |
-35.720 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
0.960 |
0.000 |
0.000 |
| y |
0.000 |
-0.798 |
0.000 |
| z |
0.000 |
0.000 |
-0.163 |
|
| Polar |
|---|
| 3z2-r2 | -0.325 |
|---|
| x2-y2 | 1.172 |
|---|
| xy | 0.000 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
8.019 |
0.000 |
0.000 |
| y |
0.000 |
10.738 |
0.000 |
| z |
0.000 |
0.000 |
8.410 |
<r2> (average value of r
2) Å
2
| <r2> |
0.000 |
| (<r2>)1/2 |
0.000 |