Jump to
S1C2
Energy calculated at BLYP/cc-pVDZ
| | hartrees |
|---|
| Energy at 0K | -578.653504 |
| Energy at 298.15K | -578.660999 |
| Nuclear repulsion energy | 155.775842 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at BLYP/cc-pVDZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3024 |
3029 |
29.60 |
|
|
|
| 2 |
A' |
2989 |
2994 |
27.82 |
|
|
|
| 3 |
A' |
2962 |
2967 |
12.19 |
|
|
|
| 4 |
A' |
2950 |
2955 |
24.30 |
|
|
|
| 5 |
A' |
1444 |
1446 |
3.06 |
|
|
|
| 6 |
A' |
1427 |
1429 |
0.61 |
|
|
|
| 7 |
A' |
1422 |
1424 |
0.66 |
|
|
|
| 8 |
A' |
1357 |
1359 |
0.32 |
|
|
|
| 9 |
A' |
1313 |
1315 |
2.32 |
|
|
|
| 10 |
A' |
1222 |
1224 |
32.49 |
|
|
|
| 11 |
A' |
1078 |
1080 |
0.37 |
|
|
|
| 12 |
A' |
1001 |
1003 |
1.37 |
|
|
|
| 13 |
A' |
878 |
879 |
12.30 |
|
|
|
| 14 |
A' |
687 |
688 |
40.85 |
|
|
|
| 15 |
A' |
345 |
345 |
3.92 |
|
|
|
| 16 |
A' |
225 |
226 |
2.04 |
|
|
|
| 17 |
A" |
3050 |
3055 |
28.57 |
|
|
|
| 18 |
A" |
3017 |
3022 |
33.96 |
|
|
|
| 19 |
A" |
2992 |
2997 |
0.35 |
|
|
|
| 20 |
A" |
1429 |
1431 |
5.73 |
|
|
|
| 21 |
A" |
1271 |
1273 |
0.00 |
|
|
|
| 22 |
A" |
1194 |
1196 |
0.15 |
|
|
|
| 23 |
A" |
1038 |
1039 |
1.01 |
|
|
|
| 24 |
A" |
841 |
843 |
0.14 |
|
|
|
| 25 |
A" |
740 |
741 |
3.32 |
|
|
|
| 26 |
A" |
225 |
226 |
0.03 |
|
|
|
| 27 |
A" |
115 |
115 |
1.24 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 20116.2 cm
-1
Scaled (by 1.0016) Zero Point Vibrational Energy (zpe) 20148.4 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at BLYP/cc-pVDZ
Point Group is Cs
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.000 |
0.592 |
0.000 |
| C2 |
0.934 |
-0.622 |
0.000 |
| C3 |
2.418 |
-0.192 |
0.000 |
| Cl4 |
-1.778 |
0.096 |
0.000 |
| H5 |
0.135 |
1.221 |
0.901 |
| H6 |
0.135 |
1.221 |
-0.901 |
| H7 |
0.717 |
-1.248 |
-0.891 |
| H8 |
0.717 |
-1.248 |
0.891 |
| H9 |
3.083 |
-1.078 |
0.000 |
| H10 |
2.667 |
0.414 |
-0.896 |
| H11 |
2.667 |
0.414 |
0.896 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
C3 |
Cl4 |
H5 |
H6 |
H7 |
H8 |
H9 |
H10 |
H11 |
| C1 | | 1.5322 | 2.5416 | 1.8463 | 1.1067 | 1.1067 | 2.1663 | 2.1663 | 3.5066 | 2.8191 | 2.8191 |
C2 | 1.5322 | | 1.5445 | 2.8061 | 2.2016 | 2.2016 | 1.1101 | 1.1101 | 2.1967 | 2.2090 | 2.2090 | C3 | 2.5416 | 1.5445 | | 4.2058 | 2.8314 | 2.8314 | 2.1914 | 2.1914 | 1.1086 | 1.1103 | 1.1103 | Cl4 | 1.8463 | 2.8061 | 4.2058 | | 2.3955 | 2.3955 | 2.9705 | 2.9705 | 5.0012 | 4.5459 | 4.5459 | H5 | 1.1067 | 2.2016 | 2.8314 | 2.3955 | | 1.8014 | 3.1052 | 2.5364 | 3.8456 | 3.2076 | 2.6571 | H6 | 1.1067 | 2.2016 | 2.8314 | 2.3955 | 1.8014 | | 2.5364 | 3.1052 | 3.8456 | 2.6571 | 3.2076 | H7 | 2.1663 | 1.1101 | 2.1914 | 2.9705 | 3.1052 | 2.5364 | | 1.7814 | 2.5342 | 2.5627 | 3.1241 | H8 | 2.1663 | 1.1101 | 2.1914 | 2.9705 | 2.5364 | 3.1052 | 1.7814 | | 2.5342 | 3.1241 | 2.5627 | H9 | 3.5066 | 2.1967 | 1.1086 | 5.0012 | 3.8456 | 3.8456 | 2.5342 | 2.5342 | | 1.7902 | 1.7902 | H10 | 2.8191 | 2.2090 | 1.1103 | 4.5459 | 3.2076 | 2.6571 | 2.5627 | 3.1241 | 1.7902 | | 1.7923 | H11 | 2.8191 | 2.2090 | 1.1103 | 4.5459 | 2.6571 | 3.2076 | 3.1241 | 2.5627 | 1.7902 | 1.7923 | |
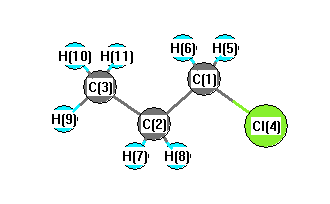 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
C3 |
111.393 |
|
C1 |
C2 |
H7 |
109.098 |
| C1 |
C2 |
H8 |
109.098 |
|
C2 |
C1 |
Cl4 |
111.981 |
| C2 |
C1 |
H5 |
112.075 |
|
C2 |
C1 |
H6 |
112.075 |
| C2 |
C3 |
H9 |
110.709 |
|
C2 |
C3 |
H10 |
111.578 |
| C2 |
C3 |
H11 |
111.578 |
|
C3 |
C2 |
H7 |
110.207 |
| C3 |
C2 |
H8 |
110.207 |
|
Cl4 |
C1 |
H5 |
105.685 |
| Cl4 |
C1 |
H6 |
105.685 |
|
H5 |
C1 |
H6 |
108.941 |
| H7 |
C2 |
H8 |
106.714 |
|
H9 |
C3 |
H10 |
107.573 |
| H9 |
C3 |
H11 |
107.573 |
|
H10 |
C3 |
H11 |
107.634 |
Electronic energy levels
Electronic state
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at BLYP/cc-pVDZ
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.030 |
|
|
|
| 2 |
C |
0.024 |
|
|
|
| 3 |
C |
0.001 |
|
|
|
| 4 |
Cl |
-0.153 |
|
|
|
| 5 |
H |
0.050 |
|
|
|
| 6 |
H |
0.050 |
|
|
|
| 7 |
H |
0.012 |
|
|
|
| 8 |
H |
0.012 |
|
|
|
| 9 |
H |
0.012 |
|
|
|
| 10 |
H |
0.012 |
|
|
|
| 11 |
H |
0.012 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
2.151 |
0.298 |
0.000 |
2.172 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-35.352 |
0.081 |
0.000 |
| y |
0.081 |
-32.620 |
0.000 |
| z |
0.000 |
0.000 |
-32.825 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-2.629 |
0.081 |
0.000 |
| y |
0.081 |
1.469 |
0.000 |
| z |
0.000 |
0.000 |
1.160 |
|
| Polar |
|---|
| 3z2-r2 | 2.321 |
|---|
| x2-y2 | -2.732 |
|---|
| xy | 0.081 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
9.046 |
0.055 |
0.000 |
| y |
0.055 |
5.934 |
0.000 |
| z |
0.000 |
0.000 |
5.530 |
<r2> (average value of r
2) Å
2
| <r2> |
157.070 |
| (<r2>)1/2 |
12.533 |
Jump to
S1C1
Energy calculated at BLYP/cc-pVDZ
| | hartrees |
|---|
| Energy at 0K | -578.653586 |
| Energy at 298.15K | -578.661207 |
| Nuclear repulsion energy | 159.533612 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at BLYP/cc-pVDZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3055 |
3060 |
16.74 |
|
|
|
| 2 |
A |
3037 |
3042 |
20.68 |
|
|
|
| 3 |
A |
3020 |
3025 |
39.65 |
|
|
|
| 4 |
A |
2996 |
3001 |
24.79 |
|
|
|
| 5 |
A |
2978 |
2982 |
8.11 |
|
|
|
| 6 |
A |
2955 |
2959 |
22.02 |
|
|
|
| 7 |
A |
2928 |
2933 |
25.55 |
|
|
|
| 8 |
A |
1441 |
1443 |
3.88 |
|
|
|
| 9 |
A |
1432 |
1434 |
6.05 |
|
|
|
| 10 |
A |
1414 |
1417 |
0.88 |
|
|
|
| 11 |
A |
1407 |
1409 |
2.12 |
|
|
|
| 12 |
A |
1360 |
1362 |
3.32 |
|
|
|
| 13 |
A |
1329 |
1331 |
3.05 |
|
|
|
| 14 |
A |
1277 |
1279 |
27.73 |
|
|
|
| 15 |
A |
1227 |
1229 |
10.75 |
|
|
|
| 16 |
A |
1185 |
1187 |
0.36 |
|
|
|
| 17 |
A |
1067 |
1069 |
1.51 |
|
|
|
| 18 |
A |
1035 |
1037 |
0.65 |
|
|
|
| 19 |
A |
1013 |
1015 |
2.74 |
|
|
|
| 20 |
A |
873 |
874 |
6.65 |
|
|
|
| 21 |
A |
834 |
836 |
5.49 |
|
|
|
| 22 |
A |
768 |
769 |
15.18 |
|
|
|
| 23 |
A |
608 |
609 |
25.63 |
|
|
|
| 24 |
A |
409 |
409 |
2.06 |
|
|
|
| 25 |
A |
290 |
290 |
0.83 |
|
|
|
| 26 |
A |
208 |
209 |
1.48 |
|
|
|
| 27 |
A |
135 |
135 |
0.84 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 20138.3 cm
-1
Scaled (by 1.0016) Zero Point Vibrational Energy (zpe) 20170.5 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at BLYP/cc-pVDZ
Point Group is C1
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.168 |
0.897 |
0.310 |
| C2 |
-1.169 |
0.563 |
-0.359 |
| C3 |
-1.837 |
-0.729 |
0.145 |
| Cl4 |
1.485 |
-0.349 |
-0.068 |
| H5 |
0.576 |
1.863 |
-0.040 |
| H6 |
0.090 |
0.914 |
1.415 |
| H7 |
-1.838 |
1.434 |
-0.167 |
| H8 |
-1.022 |
0.520 |
-1.459 |
| H9 |
-2.807 |
-0.897 |
-0.365 |
| H10 |
-1.193 |
-1.611 |
-0.046 |
| H11 |
-2.033 |
-0.682 |
1.237 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
C3 |
Cl4 |
H5 |
H6 |
H7 |
H8 |
H9 |
H10 |
H11 |
| C1 | | 1.5322 | 2.5871 | 1.8524 | 1.1053 | 1.1069 | 2.1307 | 2.1659 | 3.5394 | 2.8756 | 2.8629 |
C2 | 1.5322 | | 1.5400 | 2.8219 | 2.1988 | 2.2030 | 1.1145 | 1.1107 | 2.1948 | 2.1968 | 2.2014 | C3 | 2.5871 | 1.5400 | | 3.3508 | 3.5461 | 2.8324 | 2.1856 | 2.1904 | 1.1086 | 1.1080 | 1.1107 | Cl4 | 1.8524 | 2.8219 | 3.3508 | | 2.3918 | 2.3956 | 3.7726 | 2.9965 | 4.3376 | 2.9608 | 3.7670 | H5 | 1.1053 | 2.1988 | 3.5461 | 2.3918 | | 1.8038 | 2.4548 | 2.5241 | 4.3781 | 3.8980 | 3.8617 | H6 | 1.1069 | 2.2030 | 2.8324 | 2.3956 | 1.8038 | | 2.5472 | 3.1065 | 3.8517 | 3.1857 | 2.6611 | H7 | 2.1307 | 1.1145 | 2.1856 | 3.7726 | 2.4548 | 2.5472 | | 1.7808 | 2.5323 | 3.1145 | 2.5471 | H8 | 2.1659 | 1.1107 | 2.1904 | 2.9965 | 2.5241 | 3.1065 | 1.7808 | | 2.5284 | 2.5626 | 3.1205 | H9 | 3.5394 | 2.1948 | 1.1086 | 4.3376 | 4.3781 | 3.8517 | 2.5323 | 2.5284 | | 1.7934 | 1.7922 | H10 | 2.8756 | 2.1968 | 1.1080 | 2.9608 | 3.8980 | 3.1857 | 3.1145 | 2.5626 | 1.7934 | | 1.7923 | H11 | 2.8629 | 2.2014 | 1.1107 | 3.7670 | 3.8617 | 2.6611 | 2.5471 | 3.1205 | 1.7922 | 1.7923 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
C3 |
114.722 |
|
C1 |
C2 |
H7 |
106.152 |
| C1 |
C2 |
H8 |
109.032 |
|
C2 |
C1 |
Cl4 |
112.628 |
| C2 |
C1 |
H5 |
111.933 |
|
C2 |
C1 |
H6 |
112.176 |
| C2 |
C3 |
H9 |
110.867 |
|
C2 |
C3 |
H10 |
111.064 |
| C2 |
C3 |
H11 |
111.269 |
|
C3 |
C2 |
H7 |
109.805 |
| C3 |
C2 |
H8 |
110.406 |
|
Cl4 |
C1 |
H5 |
105.110 |
| Cl4 |
C1 |
H6 |
105.298 |
|
H5 |
C1 |
H6 |
109.250 |
| H7 |
C2 |
H8 |
106.315 |
|
H9 |
C3 |
H10 |
108.005 |
| H9 |
C3 |
H11 |
107.712 |
|
H10 |
C3 |
H11 |
107.772 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at BLYP/cc-pVDZ
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.019 |
|
|
|
| 2 |
C |
0.010 |
|
|
|
| 3 |
C |
0.013 |
|
|
|
| 4 |
Cl |
-0.155 |
|
|
|
| 5 |
H |
0.047 |
|
|
|
| 6 |
H |
0.050 |
|
|
|
| 7 |
H |
0.004 |
|
|
|
| 8 |
H |
0.011 |
|
|
|
| 9 |
H |
0.007 |
|
|
|
| 10 |
H |
0.025 |
|
|
|
| 11 |
H |
0.007 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
-1.633 |
1.223 |
0.275 |
2.059 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-34.710 |
0.208 |
0.133 |
| y |
0.208 |
-31.987 |
0.402 |
| z |
0.133 |
0.402 |
-32.862 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-2.286 |
0.208 |
0.133 |
| y |
0.208 |
1.800 |
0.402 |
| z |
0.133 |
0.402 |
0.486 |
|
| Polar |
|---|
| 3z2-r2 | 0.972 |
|---|
| x2-y2 | -2.724 |
|---|
| xy | 0.208 |
|---|
| xz | 0.133 |
|---|
| yz | 0.402 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
7.908 |
-0.553 |
-0.101 |
| y |
-0.553 |
6.693 |
0.140 |
| z |
-0.101 |
0.140 |
5.706 |
<r2> (average value of r
2) Å
2
| <r2> |
134.922 |
| (<r2>)1/2 |
11.616 |