Jump to
S1C2
Energy calculated at BLYP/cc-pVDZ
| | hartrees |
|---|
| Energy at 0K | -323.559545 |
| Energy at 298.15K | |
| HF Energy | -323.559545 |
| Nuclear repulsion energy | 229.325603 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at BLYP/cc-pVDZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3026 |
3031 |
31.51 |
|
|
|
| 2 |
A' |
2956 |
2961 |
36.96 |
|
|
|
| 3 |
A' |
2950 |
2955 |
23.88 |
|
|
|
| 4 |
A' |
2923 |
2928 |
30.64 |
|
|
|
| 5 |
A' |
1697 |
1699 |
277.29 |
|
|
|
| 6 |
A' |
1449 |
1452 |
6.44 |
|
|
|
| 7 |
A' |
1436 |
1439 |
1.83 |
|
|
|
| 8 |
A' |
1426 |
1428 |
2.65 |
|
|
|
| 9 |
A' |
1362 |
1364 |
1.69 |
|
|
|
| 10 |
A' |
1346 |
1348 |
17.35 |
|
|
|
| 11 |
A' |
1277 |
1279 |
24.82 |
|
|
|
| 12 |
A' |
1111 |
1113 |
1.88 |
|
|
|
| 13 |
A' |
1019 |
1021 |
4.83 |
|
|
|
| 14 |
A' |
989 |
990 |
19.74 |
|
|
|
| 15 |
A' |
895 |
896 |
40.59 |
|
|
|
| 16 |
A' |
750 |
751 |
286.36 |
|
|
|
| 17 |
A' |
560 |
561 |
105.27 |
|
|
|
| 18 |
A' |
347 |
348 |
0.60 |
|
|
|
| 19 |
A' |
323 |
323 |
12.03 |
|
|
|
| 20 |
A' |
145 |
145 |
0.23 |
|
|
|
| 21 |
A" |
3017 |
3021 |
67.13 |
|
|
|
| 22 |
A" |
2990 |
2995 |
12.55 |
|
|
|
| 23 |
A" |
2962 |
2966 |
16.48 |
|
|
|
| 24 |
A" |
1434 |
1436 |
5.36 |
|
|
|
| 25 |
A" |
1269 |
1271 |
0.02 |
|
|
|
| 26 |
A" |
1210 |
1212 |
0.17 |
|
|
|
| 27 |
A" |
1128 |
1130 |
0.54 |
|
|
|
| 28 |
A" |
864 |
866 |
1.12 |
|
|
|
| 29 |
A" |
746 |
747 |
2.38 |
|
|
|
| 30 |
A" |
238 |
239 |
0.00 |
|
|
|
| 31 |
A" |
205 |
206 |
0.84 |
|
|
|
| 32 |
A" |
95 |
95 |
0.97 |
|
|
|
| 33 |
A" |
82i |
82i |
0.20 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 22030.8 cm
-1
Scaled (by 1.0016) Zero Point Vibrational Energy (zpe) 22066.0 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at BLYP/cc-pVDZ
Point Group is Cs
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
-1.885 |
2.304 |
0.000 |
| C2 |
-1.517 |
0.807 |
0.000 |
| C3 |
0.000 |
0.583 |
0.000 |
| O4 |
0.234 |
-0.855 |
0.000 |
| N5 |
1.718 |
-1.118 |
0.000 |
| O6 |
1.929 |
-2.283 |
0.000 |
| H7 |
-2.985 |
2.441 |
0.000 |
| H8 |
-1.483 |
2.823 |
0.896 |
| H9 |
-1.483 |
2.823 |
-0.896 |
| H10 |
-1.951 |
0.303 |
-0.890 |
| H11 |
-1.951 |
0.303 |
0.890 |
| H12 |
0.470 |
1.037 |
0.900 |
| H13 |
0.470 |
1.037 |
-0.900 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
C3 |
O4 |
N5 |
O6 |
H7 |
H8 |
H9 |
H10 |
H11 |
H12 |
H13 |
| C1 | | 1.5418 | 2.5531 | 3.8036 | 4.9692 | 5.9657 | 1.1082 | 1.1104 | 1.1104 | 2.1905 | 2.1905 | 2.8222 | 2.8222 |
C2 | 1.5418 | | 1.5331 | 2.4134 | 3.7639 | 4.6281 | 2.1967 | 2.2061 | 2.2061 | 1.1108 | 1.1108 | 2.1933 | 2.1933 | C3 | 2.5531 | 1.5331 | | 1.4560 | 2.4171 | 3.4543 | 3.5162 | 2.8322 | 2.8322 | 2.1622 | 2.1622 | 1.1125 | 1.1125 | O4 | 3.8036 | 2.4134 | 1.4560 | | 1.5073 | 2.2167 | 4.6065 | 4.1561 | 4.1561 | 2.6277 | 2.6277 | 2.1079 | 2.1079 | N5 | 4.9692 | 3.7639 | 2.4171 | 1.5073 | | 1.1844 | 5.8975 | 5.1554 | 5.1554 | 4.0337 | 4.0337 | 2.6475 | 2.6475 | O6 | 5.9657 | 4.6281 | 3.4543 | 2.2167 | 1.1844 | | 6.8162 | 6.2060 | 6.2060 | 4.7468 | 4.7468 | 3.7363 | 3.7363 | H7 | 1.1082 | 2.1967 | 3.5162 | 4.6065 | 5.8975 | 6.8162 | | 1.7898 | 1.7898 | 2.5356 | 2.5356 | 3.8367 | 3.8367 | H8 | 1.1104 | 2.2061 | 2.8322 | 4.1561 | 5.1554 | 6.2060 | 1.7898 | | 1.7916 | 3.1230 | 2.5622 | 2.6469 | 3.1987 | H9 | 1.1104 | 2.2061 | 2.8322 | 4.1561 | 5.1554 | 6.2060 | 1.7898 | 1.7916 | | 2.5622 | 3.1230 | 3.1987 | 2.6469 | H10 | 2.1905 | 1.1108 | 2.1622 | 2.6277 | 4.0337 | 4.7468 | 2.5356 | 3.1230 | 2.5622 | | 1.7798 | 3.0988 | 2.5296 | H11 | 2.1905 | 1.1108 | 2.1622 | 2.6277 | 4.0337 | 4.7468 | 2.5356 | 2.5622 | 3.1230 | 1.7798 | | 2.5296 | 3.0988 | H12 | 2.8222 | 2.1933 | 1.1125 | 2.1079 | 2.6475 | 3.7363 | 3.8367 | 2.6469 | 3.1987 | 3.0988 | 2.5296 | | 1.8002 | H13 | 2.8222 | 2.1933 | 1.1125 | 2.1079 | 2.6475 | 3.7363 | 3.8367 | 3.1987 | 2.6469 | 2.5296 | 3.0988 | 1.8002 | |
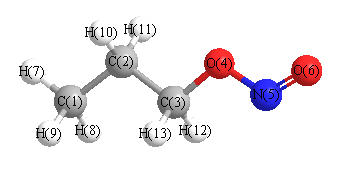 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
C3 |
112.255 |
|
C1 |
C2 |
H10 |
110.282 |
| C1 |
C2 |
H11 |
110.282 |
|
C2 |
C1 |
H7 |
110.921 |
| C2 |
C1 |
H8 |
111.526 |
|
C2 |
C1 |
H9 |
111.526 |
| C2 |
C3 |
O4 |
107.657 |
|
C2 |
C3 |
H12 |
110.998 |
| C2 |
C3 |
H13 |
110.998 |
|
C3 |
C2 |
H10 |
108.677 |
| C3 |
C2 |
H11 |
108.677 |
|
C3 |
O4 |
N5 |
109.296 |
| O4 |
C3 |
H12 |
109.582 |
|
O4 |
C3 |
H13 |
109.582 |
| O4 |
N5 |
O6 |
110.305 |
|
H7 |
C1 |
H8 |
107.557 |
| H7 |
C1 |
H9 |
107.557 |
|
H8 |
C1 |
H9 |
107.555 |
| H10 |
C2 |
H11 |
106.479 |
|
H12 |
C3 |
H13 |
108.015 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at BLYP/cc-pVDZ
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
0.001 |
|
|
|
| 2 |
C |
-0.013 |
|
|
|
| 3 |
C |
0.173 |
|
|
|
| 4 |
O |
-0.247 |
|
|
|
| 5 |
N |
0.121 |
|
|
|
| 6 |
O |
-0.091 |
|
|
|
| 7 |
H |
0.010 |
|
|
|
| 8 |
H |
0.011 |
|
|
|
| 9 |
H |
0.011 |
|
|
|
| 10 |
H |
0.007 |
|
|
|
| 11 |
H |
0.007 |
|
|
|
| 12 |
H |
0.005 |
|
|
|
| 13 |
H |
0.005 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
-0.858 |
1.927 |
0.000 |
2.109 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-36.917 |
0.703 |
0.000 |
| y |
0.703 |
-38.777 |
0.000 |
| z |
0.000 |
0.000 |
-34.989 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-0.034 |
0.703 |
0.000 |
| y |
0.703 |
-2.824 |
0.000 |
| z |
0.000 |
0.000 |
2.859 |
|
| Polar |
|---|
| 3z2-r2 | 5.717 |
|---|
| x2-y2 | 1.860 |
|---|
| xy | 0.703 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
8.852 |
-2.647 |
0.000 |
| y |
-2.647 |
9.202 |
0.000 |
| z |
0.000 |
0.000 |
5.754 |
<r2> (average value of r
2) Å
2
| <r2> |
253.514 |
| (<r2>)1/2 |
15.922 |
Jump to
S1C1
Energy calculated at BLYP/cc-pVDZ
| | hartrees |
|---|
| Energy at 0K | -323.562611 |
| Energy at 298.15K | -323.571352 |
| HF Energy | -323.562611 |
| Nuclear repulsion energy | 235.949197 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at BLYP/cc-pVDZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3032 |
3037 |
28.60 |
|
|
|
| 2 |
A |
3019 |
3024 |
41.21 |
|
|
|
| 3 |
A |
2984 |
2988 |
36.87 |
|
|
|
| 4 |
A |
2973 |
2978 |
20.62 |
|
|
|
| 5 |
A |
2954 |
2958 |
21.56 |
|
|
|
| 6 |
A |
2943 |
2948 |
34.08 |
|
|
|
| 7 |
A |
2928 |
2932 |
26.74 |
|
|
|
| 8 |
A |
1711 |
1713 |
298.90 |
|
|
|
| 9 |
A |
1440 |
1442 |
3.98 |
|
|
|
| 10 |
A |
1430 |
1433 |
5.88 |
|
|
|
| 11 |
A |
1413 |
1415 |
0.29 |
|
|
|
| 12 |
A |
1409 |
1411 |
2.59 |
|
|
|
| 13 |
A |
1360 |
1362 |
6.68 |
|
|
|
| 14 |
A |
1334 |
1336 |
12.94 |
|
|
|
| 15 |
A |
1326 |
1328 |
12.14 |
|
|
|
| 16 |
A |
1255 |
1257 |
0.56 |
|
|
|
| 17 |
A |
1231 |
1233 |
5.78 |
|
|
|
| 18 |
A |
1133 |
1134 |
3.21 |
|
|
|
| 19 |
A |
1070 |
1072 |
2.99 |
|
|
|
| 20 |
A |
1032 |
1034 |
12.14 |
|
|
|
| 21 |
A |
950 |
951 |
13.68 |
|
|
|
| 22 |
A |
898 |
899 |
25.58 |
|
|
|
| 23 |
A |
844 |
846 |
4.20 |
|
|
|
| 24 |
A |
775 |
776 |
89.48 |
|
|
|
| 25 |
A |
722 |
723 |
163.90 |
|
|
|
| 26 |
A |
521 |
522 |
56.11 |
|
|
|
| 27 |
A |
404 |
405 |
40.12 |
|
|
|
| 28 |
A |
351 |
352 |
0.13 |
|
|
|
| 29 |
A |
274 |
275 |
2.10 |
|
|
|
| 30 |
A |
235 |
235 |
1.27 |
|
|
|
| 31 |
A |
181 |
182 |
0.48 |
|
|
|
| 32 |
A |
104 |
104 |
0.93 |
|
|
|
| 33 |
A |
63 |
63 |
0.14 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 22148.5 cm
-1
Scaled (by 1.0016) Zero Point Vibrational Energy (zpe) 22184.0 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at BLYP/cc-pVDZ
Point Group is C1
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
-2.243 |
-0.928 |
0.182 |
| C2 |
-1.792 |
0.464 |
-0.313 |
| C3 |
-0.421 |
0.898 |
0.224 |
| O4 |
0.582 |
-0.029 |
-0.296 |
| N5 |
1.838 |
0.205 |
0.318 |
| O6 |
2.642 |
-0.557 |
-0.142 |
| H7 |
-3.241 |
-1.190 |
-0.223 |
| H8 |
-2.310 |
-0.956 |
1.289 |
| H9 |
-1.528 |
-1.711 |
-0.135 |
| H10 |
-1.761 |
0.485 |
-1.422 |
| H11 |
-2.527 |
1.237 |
-0.002 |
| H12 |
-0.160 |
1.923 |
-0.110 |
| H13 |
-0.391 |
0.869 |
1.333 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
C3 |
O4 |
N5 |
O6 |
H7 |
H8 |
H9 |
H10 |
H11 |
H12 |
H13 |
| C1 | | 1.5441 | 2.5794 | 3.0030 | 4.2377 | 4.9105 | 1.1080 | 1.1098 | 1.1071 | 2.1914 | 2.1905 | 3.5430 | 2.8263 |
C2 | 1.5441 | | 1.5346 | 2.4246 | 3.6935 | 4.5536 | 2.2008 | 2.2027 | 2.1978 | 1.1103 | 1.1110 | 2.1989 | 2.1995 | C3 | 2.5794 | 1.5346 | | 1.4615 | 2.3651 | 3.4113 | 3.5369 | 2.8532 | 2.8562 | 2.1620 | 2.1449 | 1.1095 | 1.1101 | O4 | 3.0030 | 2.4246 | 1.4615 | | 1.4175 | 2.1326 | 3.9959 | 3.4260 | 2.7029 | 2.6496 | 3.3697 | 2.0967 | 2.0995 | N5 | 4.2377 | 3.6935 | 2.3651 | 1.4175 | | 1.1995 | 5.2946 | 4.4159 | 3.8992 | 4.0071 | 4.4969 | 2.6696 | 2.5376 | O6 | 4.9105 | 4.5536 | 3.4113 | 2.1326 | 1.1995 | | 5.9178 | 5.1708 | 4.3270 | 4.7023 | 5.4737 | 3.7423 | 3.6621 | H7 | 1.1080 | 2.2008 | 3.5369 | 3.9959 | 5.2946 | 5.9178 | | 1.7908 | 1.7924 | 2.5372 | 2.5394 | 4.3817 | 3.8455 | H8 | 1.1098 | 2.2027 | 2.8532 | 3.4260 | 4.4159 | 5.1708 | 1.7908 | | 1.7915 | 3.1197 | 2.5542 | 3.8564 | 2.6496 | H9 | 1.1071 | 2.1978 | 2.8562 | 2.7029 | 3.8992 | 4.3270 | 1.7924 | 1.7915 | | 2.5562 | 3.1151 | 3.8831 | 3.1791 | H10 | 2.1914 | 1.1103 | 2.1620 | 2.6496 | 4.0071 | 4.7023 | 2.5372 | 3.1197 | 2.5562 | | 1.7803 | 2.5208 | 3.1013 | H11 | 2.1905 | 1.1110 | 2.1449 | 3.3697 | 4.4969 | 5.4737 | 2.5394 | 2.5542 | 3.1151 | 1.7803 | | 2.4672 | 2.5462 | H12 | 3.5430 | 2.1989 | 1.1095 | 2.0967 | 2.6696 | 3.7423 | 4.3817 | 3.8564 | 3.8831 | 2.5208 | 2.4672 | | 1.8013 | H13 | 2.8263 | 2.1995 | 1.1101 | 2.0995 | 2.5376 | 3.6621 | 3.8455 | 2.6496 | 3.1791 | 3.1013 | 2.5462 | 1.8013 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
C3 |
113.823 |
|
C1 |
C2 |
H10 |
110.226 |
| C1 |
C2 |
H11 |
110.115 |
|
C2 |
C1 |
H7 |
111.097 |
| C2 |
C1 |
H8 |
111.136 |
|
C2 |
C1 |
H9 |
110.916 |
| C2 |
C3 |
O4 |
108.020 |
|
C2 |
C3 |
H12 |
111.523 |
| C2 |
C3 |
H13 |
111.531 |
|
C3 |
C2 |
H10 |
108.592 |
| C3 |
C2 |
H11 |
107.245 |
|
C3 |
O4 |
N5 |
110.464 |
| O4 |
C3 |
H12 |
108.504 |
|
O4 |
C3 |
H13 |
108.684 |
| O4 |
N5 |
O6 |
108.869 |
|
H7 |
C1 |
H8 |
107.695 |
| H7 |
C1 |
H9 |
108.028 |
|
H8 |
C1 |
H9 |
107.820 |
| H10 |
C2 |
H11 |
106.544 |
|
H12 |
C3 |
H13 |
108.498 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at BLYP/cc-pVDZ
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
0.010 |
|
|
|
| 2 |
C |
-0.010 |
|
|
|
| 3 |
C |
0.167 |
|
|
|
| 4 |
O |
-0.234 |
|
|
|
| 5 |
N |
0.120 |
|
|
|
| 6 |
O |
-0.083 |
|
|
|
| 7 |
H |
0.006 |
|
|
|
| 8 |
H |
0.008 |
|
|
|
| 9 |
H |
0.012 |
|
|
|
| 10 |
H |
0.007 |
|
|
|
| 11 |
H |
-0.004 |
|
|
|
| 12 |
H |
0.006 |
|
|
|
| 13 |
H |
-0.004 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
1.633 |
-0.524 |
0.710 |
1.857 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-37.380 |
0.064 |
-0.673 |
| y |
0.064 |
-35.179 |
-0.712 |
| z |
-0.673 |
-0.712 |
-37.026 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-1.277 |
0.064 |
-0.673 |
| y |
0.064 |
2.024 |
-0.712 |
| z |
-0.673 |
-0.712 |
-0.746 |
|
| Polar |
|---|
| 3z2-r2 | -1.493 |
|---|
| x2-y2 | -2.200 |
|---|
| xy | 0.064 |
|---|
| xz | -0.673 |
|---|
| yz | -0.712 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
10.191 |
-1.314 |
-0.056 |
| y |
-1.314 |
7.183 |
-0.017 |
| z |
-0.056 |
-0.017 |
6.186 |
<r2> (average value of r
2) Å
2
| <r2> |
203.583 |
| (<r2>)1/2 |
14.268 |