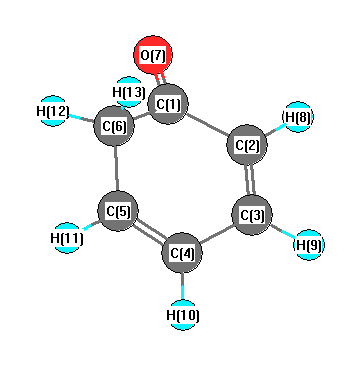
Vibrational Frequencies calculated at BLYP/cc-pVTZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A1 |
3107 |
3097 |
0.68 |
|
|
|
| 2 |
A1 |
3069 |
3060 |
12.62 |
|
|
|
| 3 |
A1 |
2906 |
2897 |
11.68 |
|
|
|
| 4 |
A1 |
1646 |
1641 |
164.50 |
|
|
|
| 5 |
A1 |
1618 |
1613 |
89.73 |
|
|
|
| 6 |
A1 |
1389 |
1385 |
20.80 |
|
|
|
| 7 |
A1 |
1382 |
1378 |
3.57 |
|
|
|
| 8 |
A1 |
1166 |
1163 |
7.65 |
|
|
|
| 9 |
A1 |
942 |
939 |
7.56 |
|
|
|
| 10 |
A1 |
854 |
851 |
9.09 |
|
|
|
| 11 |
A1 |
745 |
743 |
2.35 |
|
|
|
| 12 |
A1 |
496 |
495 |
3.14 |
|
|
|
| 13 |
A2 |
1168 |
1164 |
0.00 |
|
|
|
| 14 |
A2 |
986 |
983 |
0.00 |
|
|
|
| 15 |
A2 |
730 |
728 |
0.00 |
|
|
|
| 16 |
A2 |
354 |
353 |
0.00 |
|
|
|
| 17 |
B1 |
2903 |
2894 |
6.65 |
|
|
|
| 18 |
B1 |
1001 |
998 |
0.00 |
|
|
|
| 19 |
B1 |
921 |
918 |
24.17 |
|
|
|
| 20 |
B1 |
838 |
836 |
26.68 |
|
|
|
| 21 |
B1 |
561 |
559 |
22.32 |
|
|
|
| 22 |
B1 |
311 |
310 |
2.48 |
|
|
|
| 23 |
B1 |
123 |
123 |
0.81 |
|
|
|
| 24 |
B2 |
3105 |
3096 |
21.99 |
|
|
|
| 25 |
B2 |
3068 |
3059 |
17.04 |
|
|
|
| 26 |
B2 |
1589 |
1585 |
2.77 |
|
|
|
| 27 |
B2 |
1371 |
1367 |
24.14 |
|
|
|
| 28 |
B2 |
1332 |
1328 |
0.10 |
|
|
|
| 29 |
B2 |
1233 |
1229 |
26.27 |
|
|
|
| 30 |
B2 |
1112 |
1108 |
12.01 |
|
|
|
| 31 |
B2 |
963 |
960 |
6.92 |
|
|
|
| 32 |
B2 |
564 |
562 |
2.07 |
|
|
|
| 33 |
B2 |
443 |
441 |
11.66 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 21996.5 cm
-1
Scaled (by 0.997) Zero Point Vibrational Energy (zpe) 21930.5 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at BLYP/cc-pVTZ
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
0.210 |
|
|
|
| 2 |
C |
-0.096 |
|
|
|
| 3 |
C |
-0.137 |
|
|
|
| 4 |
C |
-0.137 |
|
|
|
| 5 |
C |
-0.083 |
|
|
|
| 6 |
C |
-0.083 |
|
|
|
| 7 |
O |
-0.292 |
|
|
|
| 8 |
H |
0.104 |
|
|
|
| 9 |
H |
0.104 |
|
|
|
| 10 |
H |
0.107 |
|
|
|
| 11 |
H |
0.107 |
|
|
|
| 12 |
H |
0.098 |
|
|
|
| 13 |
H |
0.098 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
-4.548 |
4.548 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-42.349 |
0.000 |
0.000 |
| y |
0.000 |
-37.160 |
0.000 |
| z |
0.000 |
0.000 |
-45.935 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-0.802 |
0.000 |
0.000 |
| y |
0.000 |
6.982 |
0.000 |
| z |
0.000 |
0.000 |
-6.180 |
|
| Polar |
|---|
| 3z2-r2 | -12.361 |
|---|
| x2-y2 | -5.190 |
|---|
| xy | 0.000 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
5.853 |
0.000 |
0.000 |
| y |
0.000 |
10.694 |
0.000 |
| z |
0.000 |
0.000 |
14.840 |
<r2> (average value of r
2) Å
2
| <r2> |
189.656 |
| (<r2>)1/2 |
13.772 |