Jump to
S1C2
Vibrational Frequencies calculated at BLYP/6-311G*
Geometric Data calculated at BLYP/6-311G*
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Jump to
S1C1
Energy calculated at BLYP/6-311G*
| | hartrees |
|---|
| Energy at 0K | -235.750312 |
| Energy at 298.15K | -235.762332 |
| HF Energy | -235.750312 |
| Nuclear repulsion energy | 224.452841 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at BLYP/6-311G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3029 |
3022 |
92.78 |
|
|
|
| 2 |
A |
3014 |
3007 |
21.54 |
|
|
|
| 3 |
A |
3008 |
3001 |
53.14 |
|
|
|
| 4 |
A |
3007 |
2999 |
2.31 |
|
|
|
| 5 |
A |
3005 |
2997 |
73.75 |
|
|
|
| 6 |
A |
2971 |
2964 |
21.43 |
|
|
|
| 7 |
A |
2968 |
2961 |
34.01 |
|
|
|
| 8 |
A |
2953 |
2945 |
33.56 |
|
|
|
| 9 |
A |
2948 |
2941 |
30.51 |
|
|
|
| 10 |
A |
2939 |
2931 |
25.80 |
|
|
|
| 11 |
A |
2935 |
2928 |
47.92 |
|
|
|
| 12 |
A |
2910 |
2903 |
32.56 |
|
|
|
| 13 |
A |
1676 |
1672 |
0.69 |
|
|
|
| 14 |
A |
1490 |
1487 |
6.66 |
|
|
|
| 15 |
A |
1482 |
1479 |
6.66 |
|
|
|
| 16 |
A |
1476 |
1472 |
10.89 |
|
|
|
| 17 |
A |
1474 |
1470 |
0.77 |
|
|
|
| 18 |
A |
1462 |
1458 |
6.99 |
|
|
|
| 19 |
A |
1457 |
1453 |
2.10 |
|
|
|
| 20 |
A |
1391 |
1387 |
1.22 |
|
|
|
| 21 |
A |
1389 |
1386 |
2.63 |
|
|
|
| 22 |
A |
1348 |
1345 |
1.51 |
|
|
|
| 23 |
A |
1322 |
1319 |
1.06 |
|
|
|
| 24 |
A |
1313 |
1309 |
0.39 |
|
|
|
| 25 |
A |
1296 |
1292 |
0.06 |
|
|
|
| 26 |
A |
1269 |
1266 |
6.68 |
|
|
|
| 27 |
A |
1231 |
1228 |
0.32 |
|
|
|
| 28 |
A |
1149 |
1146 |
0.05 |
|
|
|
| 29 |
A |
1082 |
1080 |
0.51 |
|
|
|
| 30 |
A |
1054 |
1052 |
0.40 |
|
|
|
| 31 |
A |
1041 |
1038 |
0.25 |
|
|
|
| 32 |
A |
1026 |
1024 |
11.35 |
|
|
|
| 33 |
A |
994 |
992 |
0.99 |
|
|
|
| 34 |
A |
968 |
966 |
33.56 |
|
|
|
| 35 |
A |
906 |
904 |
4.19 |
|
|
|
| 36 |
A |
872 |
870 |
2.50 |
|
|
|
| 37 |
A |
853 |
851 |
0.43 |
|
|
|
| 38 |
A |
753 |
751 |
0.80 |
|
|
|
| 39 |
A |
728 |
726 |
1.92 |
|
|
|
| 40 |
A |
520 |
519 |
0.15 |
|
|
|
| 41 |
A |
375 |
374 |
1.23 |
|
|
|
| 42 |
A |
304 |
304 |
0.85 |
|
|
|
| 43 |
A |
277 |
277 |
1.55 |
|
|
|
| 44 |
A |
237 |
236 |
0.05 |
|
|
|
| 45 |
A |
200 |
199 |
0.67 |
|
|
|
| 46 |
A |
139 |
139 |
0.32 |
|
|
|
| 47 |
A |
95 |
95 |
0.05 |
|
|
|
| 48 |
A |
76 |
76 |
0.22 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 35205.2 cm
-1
Scaled (by 0.9975) Zero Point Vibrational Energy (zpe) 35117.2 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at BLYP/6-311G*
Point Group is C1
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
3.214 |
0.216 |
-0.158 |
| C2 |
1.822 |
-0.364 |
-0.222 |
| C3 |
0.751 |
0.103 |
0.438 |
| C4 |
-0.645 |
-0.475 |
0.374 |
| C5 |
-1.702 |
0.524 |
-0.170 |
| C6 |
-3.130 |
-0.056 |
-0.192 |
| H7 |
3.261 |
1.096 |
0.500 |
| H8 |
3.942 |
-0.524 |
0.216 |
| H9 |
3.570 |
0.522 |
-1.157 |
| H10 |
1.700 |
-1.245 |
-0.865 |
| H11 |
0.872 |
0.985 |
1.082 |
| H12 |
-0.958 |
-0.796 |
1.385 |
| H13 |
-0.645 |
-1.384 |
-0.251 |
| H14 |
-1.409 |
0.836 |
-1.186 |
| H15 |
-1.685 |
1.440 |
0.446 |
| H16 |
-3.854 |
0.675 |
-0.582 |
| H17 |
-3.190 |
-0.954 |
-0.827 |
| H18 |
-3.462 |
-0.344 |
0.818 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
C3 |
C4 |
C5 |
C6 |
H7 |
H8 |
H9 |
H10 |
H11 |
H12 |
H13 |
H14 |
H15 |
H16 |
H17 |
H18 |
| C1 | | 1.5098 | 2.5374 | 3.9568 | 4.9257 | 6.3502 | 1.0998 | 1.1033 | 1.1032 | 2.2204 | 2.7599 | 4.5619 | 4.1787 | 4.7760 | 5.0863 | 7.0958 | 6.5442 | 6.7709 |
C2 | 1.5098 | | 1.3419 | 2.5403 | 3.6341 | 4.9614 | 2.1733 | 2.1708 | 2.1711 | 1.0981 | 2.1028 | 3.2396 | 2.6694 | 3.5781 | 4.0000 | 5.7812 | 5.0823 | 5.3856 | C3 | 2.5374 | 1.3419 | | 1.5119 | 2.5614 | 3.9345 | 2.7005 | 3.2599 | 3.2662 | 2.1015 | 1.0989 | 2.1499 | 2.1523 | 2.7993 | 2.7789 | 4.7508 | 4.2711 | 4.2536 | C4 | 3.9568 | 2.5403 | 1.5119 | | 1.5519 | 2.5824 | 4.2121 | 4.5901 | 4.5937 | 2.7617 | 2.2211 | 1.1065 | 1.1033 | 2.1752 | 2.1801 | 3.5399 | 2.8540 | 2.8550 | C5 | 4.9257 | 3.6341 | 2.5614 | 1.5519 | | 1.5415 | 5.0403 | 5.7531 | 5.3633 | 3.8966 | 2.8986 | 2.1710 | 2.1831 | 1.1026 | 1.1037 | 2.1966 | 2.1975 | 2.1974 | C6 | 6.3502 | 4.9614 | 3.9345 | 2.5824 | 1.5415 | | 6.5306 | 7.0991 | 6.7936 | 5.0192 | 4.3265 | 2.7845 | 2.8188 | 2.1787 | 2.1750 | 1.1004 | 1.1014 | 1.1014 | H7 | 1.0998 | 2.1733 | 2.7005 | 4.2121 | 5.0403 | 6.5306 | | 1.7802 | 1.7803 | 3.1279 | 2.4617 | 4.7074 | 4.6872 | 4.9711 | 4.9586 | 7.2091 | 6.8973 | 6.8833 | H8 | 1.1033 | 2.1708 | 3.2599 | 4.5901 | 5.7531 | 7.0991 | 1.7802 | | 1.7656 | 2.5919 | 3.5289 | 5.0444 | 4.6898 | 5.6957 | 5.9646 | 7.9278 | 7.2203 | 7.4309 | H9 | 1.1032 | 2.1711 | 3.2662 | 4.5937 | 5.3633 | 6.7936 | 1.7803 | 1.7656 | | 2.5895 | 3.5365 | 5.3569 | 4.7134 | 4.9883 | 5.5704 | 7.4475 | 6.9265 | 7.3554 | H10 | 2.2204 | 1.0981 | 2.1015 | 2.7617 | 3.8966 | 5.0192 | 3.1279 | 2.5919 | 2.5895 | | 3.0742 | 3.5112 | 2.4274 | 3.7542 | 4.5153 | 5.8828 | 4.8982 | 5.5037 | H11 | 2.7599 | 2.1028 | 1.0989 | 2.2211 | 2.8986 | 4.3265 | 2.4617 | 3.5289 | 3.5365 | 3.0742 | | 2.5709 | 3.1127 | 3.2192 | 2.6740 | 5.0198 | 4.8885 | 4.5410 | H12 | 4.5619 | 3.2396 | 2.1499 | 1.1065 | 2.1710 | 2.7845 | 4.7074 | 5.0444 | 5.3569 | 3.5112 | 2.5709 | | 1.7667 | 3.0783 | 2.5318 | 3.7979 | 3.1468 | 2.6076 | H13 | 4.1787 | 2.6694 | 2.1523 | 1.1033 | 2.1831 | 2.8188 | 4.6872 | 4.6898 | 4.7134 | 2.4274 | 3.1127 | 1.7667 | | 2.5274 | 3.0896 | 3.8275 | 2.6449 | 3.1881 | H14 | 4.7760 | 3.5781 | 2.7993 | 2.1752 | 1.1026 | 2.1787 | 4.9711 | 5.6957 | 4.9883 | 3.7542 | 3.2192 | 3.0783 | 2.5274 | | 1.7617 | 2.5238 | 2.5502 | 3.1026 | H15 | 5.0863 | 4.0000 | 2.7789 | 2.1801 | 1.1037 | 2.1750 | 4.9586 | 5.9646 | 5.5704 | 4.5153 | 2.6740 | 2.5318 | 3.0896 | 1.7617 | | 2.5190 | 3.1003 | 2.5454 | H16 | 7.0958 | 5.7812 | 4.7508 | 3.5399 | 2.1966 | 1.1004 | 7.2091 | 7.9278 | 7.4475 | 5.8828 | 5.0198 | 3.7979 | 3.8275 | 2.5238 | 2.5190 | | 1.7754 | 1.7758 | H17 | 6.5442 | 5.0823 | 4.2711 | 2.8540 | 2.1975 | 1.1014 | 6.8973 | 7.2203 | 6.9265 | 4.8982 | 4.8885 | 3.1468 | 2.6449 | 2.5502 | 3.1003 | 1.7754 | | 1.7753 | H18 | 6.7709 | 5.3856 | 4.2536 | 2.8550 | 2.1974 | 1.1014 | 6.8833 | 7.4309 | 7.3554 | 5.5037 | 4.5410 | 2.6076 | 3.1881 | 3.1026 | 2.5454 | 1.7758 | 1.7753 | |
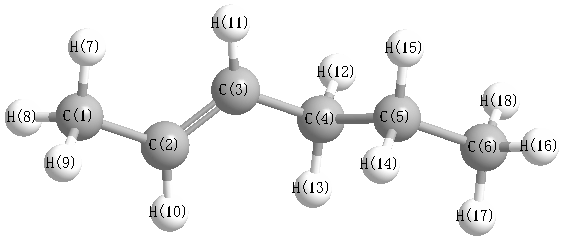 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
C3 |
125.591 |
|
C1 |
C2 |
H10 |
115.831 |
| C2 |
C1 |
H7 |
111.823 |
|
C2 |
C1 |
H8 |
111.403 |
| C2 |
C1 |
H9 |
111.433 |
|
C2 |
C3 |
C4 |
125.680 |
| C2 |
C3 |
H11 |
118.636 |
|
C3 |
C2 |
H10 |
118.577 |
| C3 |
C4 |
C5 |
113.439 |
|
C3 |
C4 |
H12 |
109.412 |
| C3 |
C4 |
H13 |
109.785 |
|
C4 |
C3 |
H11 |
115.683 |
| C4 |
C5 |
C6 |
113.189 |
|
C4 |
C5 |
H14 |
108.876 |
| C4 |
C5 |
H15 |
109.192 |
|
C5 |
C4 |
H12 |
108.332 |
| C5 |
C4 |
H13 |
109.446 |
|
C5 |
C6 |
H16 |
111.402 |
| C5 |
C6 |
H17 |
111.411 |
|
C5 |
C6 |
H18 |
111.407 |
| C6 |
C5 |
H14 |
109.859 |
|
C6 |
C5 |
H15 |
109.503 |
| H7 |
C1 |
H8 |
107.808 |
|
H7 |
C1 |
H9 |
107.828 |
| H8 |
C1 |
H9 |
106.287 |
|
H12 |
C4 |
H13 |
106.160 |
| H14 |
C5 |
H15 |
105.963 |
|
H16 |
C6 |
H17 |
107.487 |
| H16 |
C6 |
H18 |
107.515 |
|
H17 |
C6 |
H18 |
107.403 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at BLYP/6-311G*
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.618 |
|
|
|
| 2 |
C |
-0.145 |
|
|
|
| 3 |
C |
-0.135 |
|
|
|
| 4 |
C |
-0.411 |
|
|
|
| 5 |
C |
-0.369 |
|
|
|
| 6 |
C |
-0.586 |
|
|
|
| 7 |
H |
0.198 |
|
|
|
| 8 |
H |
0.202 |
|
|
|
| 9 |
H |
0.202 |
|
|
|
| 10 |
H |
0.162 |
|
|
|
| 11 |
H |
0.159 |
|
|
|
| 12 |
H |
0.192 |
|
|
|
| 13 |
H |
0.189 |
|
|
|
| 14 |
H |
0.193 |
|
|
|
| 15 |
H |
0.186 |
|
|
|
| 16 |
H |
0.197 |
|
|
|
| 17 |
H |
0.192 |
|
|
|
| 18 |
H |
0.192 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.043 |
0.036 |
-0.037 |
0.067 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-40.159 |
-0.116 |
-0.646 |
| y |
-0.116 |
-40.804 |
1.257 |
| z |
-0.646 |
1.257 |
-40.849 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
0.667 |
-0.116 |
-0.646 |
| y |
-0.116 |
-0.300 |
1.257 |
| z |
-0.646 |
1.257 |
-0.367 |
|
| Polar |
|---|
| 3z2-r2 | -0.734 |
|---|
| x2-y2 | 0.645 |
|---|
| xy | -0.116 |
|---|
| xz | -0.646 |
|---|
| yz | 1.257 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
14.805 |
-0.180 |
-0.901 |
| y |
-0.180 |
9.171 |
0.635 |
| z |
-0.901 |
0.635 |
8.888 |
<r2> (average value of r
2) Å
2
| <r2> |
301.926 |
| (<r2>)1/2 |
17.376 |