Jump to
S1C2
S1C3
Energy calculated at CID/6-31G*
| | hartrees |
|---|
| Energy at 0K | -580.256782 |
| Energy at 298.15K | |
| HF Energy | -580.076348 |
| Nuclear repulsion energy | 78.816858 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at CID/6-31G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
Ag |
2335 |
2157 |
0.00 |
|
|
|
| 2 |
Ag |
976 |
902 |
0.00 |
|
|
|
| 3 |
Ag |
612 |
565 |
0.00 |
|
|
|
| 4 |
Au |
554 |
511 |
0.00 |
|
|
|
| 5 |
B1u |
2326 |
2148 |
95.42 |
|
|
|
| 6 |
B1u |
897 |
828 |
133.38 |
|
|
|
| 7 |
B2g |
244i |
225i |
0.00 |
|
|
|
| 8 |
B2u |
2361 |
2181 |
175.17 |
|
|
|
| 9 |
B2u |
369 |
341 |
20.54 |
|
|
|
| 10 |
B3g |
2353 |
2173 |
0.00 |
|
|
|
| 11 |
B3g |
614 |
567 |
0.00 |
|
|
|
| 12 |
B3u |
538 |
497 |
2.36 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 6844.6 cm
-1
Scaled (by 0.9237) Zero Point Vibrational Energy (zpe) 6322.3 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at CID/6-31G*
Point Group is D2h
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| Si1 |
0.000 |
0.000 |
1.069 |
| Si2 |
0.000 |
0.000 |
-1.069 |
| H3 |
0.000 |
1.252 |
1.856 |
| H4 |
0.000 |
-1.252 |
1.856 |
| H5 |
0.000 |
1.252 |
-1.856 |
| H6 |
0.000 |
-1.252 |
-1.856 |
Atom - Atom Distances (Å)
| |
Si1 |
Si2 |
H3 |
H4 |
H5 |
H6 |
| Si1 | | 2.1375 | 1.4791 | 1.4791 | 3.1816 | 3.1816 |
Si2 | 2.1375 | | 3.1816 | 3.1816 | 1.4791 | 1.4791 | H3 | 1.4791 | 3.1816 | | 2.5043 | 3.7121 | 4.4779 | H4 | 1.4791 | 3.1816 | 2.5043 | | 4.4779 | 3.7121 | H5 | 3.1816 | 1.4791 | 3.7121 | 4.4779 | | 2.5043 | H6 | 3.1816 | 1.4791 | 4.4779 | 3.7121 | 2.5043 | |
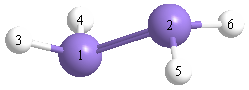 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| Si1 |
Si2 |
H5 |
122.160 |
|
Si1 |
Si2 |
H6 |
122.160 |
| Si2 |
Si1 |
H3 |
122.160 |
|
Si2 |
Si1 |
H4 |
122.160 |
| H3 |
Si1 |
H4 |
115.680 |
|
H5 |
Si2 |
H6 |
115.680 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Jump to
S1C1
S1C3
Energy calculated at CID/6-31G*
| | hartrees |
|---|
| Energy at 0K | -580.257735 |
| Energy at 298.15K | |
| HF Energy | -580.075962 |
| Nuclear repulsion energy | 78.277838 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at CID/6-31G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
Ag |
2315 |
2138 |
0.00 |
|
|
|
| 2 |
Ag |
981 |
906 |
0.00 |
|
|
|
| 3 |
Ag |
593 |
548 |
0.00 |
|
|
|
| 4 |
Ag |
318 |
293 |
0.00 |
|
|
|
| 5 |
Au |
2339 |
2161 |
202.52 |
|
|
|
| 6 |
Au |
546 |
504 |
0.04 |
|
|
|
| 7 |
Au |
358 |
330 |
21.10 |
|
|
|
| 8 |
Bg |
2330 |
2152 |
0.00 |
|
|
|
| 9 |
Bg |
632 |
584 |
0.00 |
|
|
|
| 10 |
Bu |
2307 |
2131 |
133.64 |
|
|
|
| 11 |
Bu |
936 |
864 |
187.63 |
|
|
|
| 12 |
Bu |
491 |
453 |
18.75 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 7072.2 cm
-1
Scaled (by 0.9237) Zero Point Vibrational Energy (zpe) 6532.6 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at CID/6-31G*
Point Group is C2h
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| Si1 |
0.000 |
1.081 |
0.000 |
| Si2 |
0.000 |
-1.081 |
0.000 |
| H3 |
0.380 |
1.808 |
1.236 |
| H4 |
0.380 |
1.808 |
-1.236 |
| H5 |
-0.380 |
-1.808 |
1.236 |
| H6 |
-0.380 |
-1.808 |
-1.236 |
Atom - Atom Distances (Å)
| |
Si1 |
Si2 |
H3 |
H4 |
H5 |
H6 |
| Si1 | | 2.1616 | 1.4832 | 1.4832 | 3.1648 | 3.1648 |
Si2 | 2.1616 | | 3.1648 | 3.1648 | 1.4832 | 1.4832 | H3 | 1.4832 | 3.1648 | | 2.4710 | 3.6951 | 4.4452 | H4 | 1.4832 | 3.1648 | 2.4710 | | 4.4452 | 3.6951 | H5 | 3.1648 | 1.4832 | 3.6951 | 4.4452 | | 2.4710 | H6 | 3.1648 | 1.4832 | 4.4452 | 3.6951 | 2.4710 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| Si1 |
Si2 |
H5 |
119.358 |
|
Si1 |
Si2 |
H6 |
119.358 |
| Si2 |
Si1 |
H3 |
119.358 |
|
Si2 |
Si1 |
H4 |
119.358 |
| H3 |
Si1 |
H4 |
112.815 |
|
H5 |
Si2 |
H6 |
112.815 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Jump to
S1C1
S1C2
Energy calculated at CID/6-31G*
| | hartrees |
|---|
| Energy at 0K | -580.214192 |
| Energy at 298.15K | |
| HF Energy | -580.034941 |
| Nuclear repulsion energy | 73.508223 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at CID/6-31G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A1 |
2176 |
2010 |
424.91 |
|
|
|
| 2 |
A1 |
1723 |
1592 |
0.00 |
|
|
|
| 3 |
A1 |
963 |
890 |
56.73 |
|
|
|
| 4 |
A1 |
419 |
387 |
0.44 |
|
|
|
| 5 |
A1 |
405 |
374 |
1.90 |
|
|
|
| 6 |
A2 |
1404 |
1297 |
0.00 |
|
|
|
| 7 |
A2 |
664 |
613 |
0.00 |
|
|
|
| 8 |
B1 |
1368 |
1263 |
84.32 |
|
|
|
| 9 |
B1 |
922 |
852 |
19.49 |
|
|
|
| 10 |
B2 |
2149 |
1985 |
15.95 |
|
|
|
| 11 |
B2 |
1516 |
1400 |
1822.91 |
|
|
|
| 12 |
B2 |
799 |
738 |
108.55 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 7253.0 cm
-1
Scaled (by 0.9237) Zero Point Vibrational Energy (zpe) 6699.6 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at CID/6-31G*
Point Group is C2v
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| Si1 |
0.000 |
1.306 |
-0.093 |
| Si2 |
0.000 |
-1.306 |
-0.093 |
| H3 |
-1.029 |
0.000 |
-0.119 |
| H4 |
1.029 |
0.000 |
-0.119 |
| H5 |
0.000 |
1.372 |
1.419 |
| H6 |
0.000 |
-1.372 |
1.419 |
Atom - Atom Distances (Å)
| |
Si1 |
Si2 |
H3 |
H4 |
H5 |
H6 |
| Si1 | | 2.6117 | 1.6630 | 1.6630 | 1.5136 | 3.0752 |
Si2 | 2.6117 | | 1.6630 | 1.6630 | 3.0752 | 1.5136 | H3 | 1.6630 | 1.6630 | | 2.0589 | 2.3036 | 2.3036 | H4 | 1.6630 | 1.6630 | 2.0589 | | 2.3036 | 2.3036 | H5 | 1.5136 | 3.0752 | 2.3036 | 2.3036 | | 2.7438 | H6 | 3.0752 | 1.5136 | 2.3036 | 2.3036 | 2.7438 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| Si1 |
Si2 |
H5 |
29.454 |
|
Si1 |
Si2 |
H6 |
92.500 |
| Si2 |
Si1 |
H3 |
38.258 |
|
Si2 |
Si1 |
H4 |
38.258 |
| H3 |
Si1 |
H4 |
76.489 |
|
H5 |
Si2 |
H6 |
63.046 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability