All results from a given calculation for CH2FCH2OH (2-fluoroethanol)
using model chemistry: CID/daug-cc-pVTZ
19 10 17 12 22
States and conformations
| State |
Conformation |
minimum conformation |
conformer description |
state description |
| 1 |
1 |
yes |
C1 |
1A |
Energy calculated at CID/daug-cc-pVTZ
| | hartrees |
|---|
| Energy at 0K | -253.793090 |
| Energy at 298.15K | |
| HF Energy | -253.038762 |
| Nuclear repulsion energy | 132.027491 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at CID/daug-cc-pVTZ
Geometric Data calculated at CID/daug-cc-pVTZ
Point Group is C1
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.676 |
0.569 |
0.282 |
| C2 |
-0.716 |
0.547 |
-0.279 |
| O3 |
1.448 |
-0.500 |
-0.190 |
| F4 |
-1.352 |
-0.596 |
0.156 |
| H5 |
1.169 |
1.480 |
-0.034 |
| H6 |
0.628 |
0.570 |
1.367 |
| H7 |
-1.289 |
1.400 |
0.063 |
| H8 |
-0.699 |
0.520 |
-1.360 |
| H9 |
1.022 |
-1.311 |
0.060 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
O3 |
F4 |
H5 |
H6 |
H7 |
H8 |
H9 |
| C1 | | 1.5014 | 1.3999 | 2.3427 | 1.0824 | 1.0865 | 2.1450 | 2.1425 | 1.9242 |
C2 | 1.5014 | | 2.4055 | 1.3783 | 2.1174 | 2.1252 | 1.0827 | 1.0818 | 2.5666 | O3 | 1.3999 | 2.4055 | | 2.8226 | 2.0052 | 2.0589 | 3.3410 | 2.6493 | 0.9494 | F4 | 2.3427 | 1.3783 | 2.8226 | | 3.2711 | 2.5974 | 1.9988 | 1.9923 | 2.4811 | H5 | 1.0824 | 2.1174 | 2.0052 | 3.2711 | | 1.7563 | 2.4612 | 2.4838 | 2.7961 | H6 | 1.0865 | 2.1252 | 2.0589 | 2.5974 | 1.7563 | | 2.4628 | 3.0334 | 2.3235 | H7 | 2.1450 | 1.0827 | 3.3410 | 1.9988 | 2.4612 | 2.4628 | | 1.7742 | 3.5620 | H8 | 2.1425 | 1.0818 | 2.6493 | 1.9923 | 2.4838 | 3.0334 | 1.7742 | | 2.8863 | H9 | 1.9242 | 2.5666 | 0.9494 | 2.4811 | 2.7961 | 2.3235 | 3.5620 | 2.8863 | |
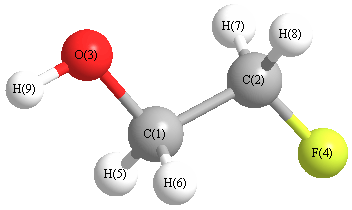 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
F4 |
108.810 |
|
C1 |
C2 |
H7 |
111.187 |
| C1 |
C2 |
H8 |
111.032 |
|
C1 |
O3 |
H9 |
108.456 |
| C2 |
C1 |
O3 |
111.968 |
|
C2 |
C1 |
H5 |
108.994 |
| C2 |
C1 |
H6 |
109.368 |
|
O3 |
C1 |
H5 |
107.072 |
| O3 |
C1 |
H6 |
111.172 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability