Jump to
S1C2
Vibrational Frequencies calculated at CISD/6-31G*
Geometric Data calculated at CISD/6-31G*
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Jump to
S1C1
Energy calculated at CISD/6-31G*
| | hartrees |
|---|
| Energy at 0K | -234.864358 |
| Energy at 298.15K | -234.878037 |
| HF Energy | -234.170048 |
| Nuclear repulsion energy | 250.660509 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at CISD/6-31G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3232 |
2992 |
118.39 |
|
|
|
| 2 |
A' |
3226 |
2987 |
21.90 |
|
|
|
| 3 |
A' |
3220 |
2981 |
22.53 |
|
|
|
| 4 |
A' |
3187 |
2951 |
19.49 |
|
|
|
| 5 |
A' |
3172 |
2937 |
12.31 |
|
|
|
| 6 |
A' |
3168 |
2933 |
9.99 |
|
|
|
| 7 |
A' |
3153 |
2919 |
26.72 |
|
|
|
| 8 |
A' |
3150 |
2916 |
26.39 |
|
|
|
| 9 |
A' |
1607 |
1487 |
4.80 |
|
|
|
| 10 |
A' |
1595 |
1477 |
2.06 |
|
|
|
| 11 |
A' |
1593 |
1475 |
5.63 |
|
|
|
| 12 |
A' |
1517 |
1404 |
8.36 |
|
|
|
| 13 |
A' |
1515 |
1403 |
0.98 |
|
|
|
| 14 |
A' |
1476 |
1367 |
1.44 |
|
|
|
| 15 |
A' |
1463 |
1354 |
7.11 |
|
|
|
| 16 |
A' |
1371 |
1270 |
1.48 |
|
|
|
| 17 |
A' |
1326 |
1228 |
0.57 |
|
|
|
| 18 |
A' |
1275 |
1180 |
0.10 |
|
|
|
| 19 |
A' |
1203 |
1113 |
0.30 |
|
|
|
| 20 |
A' |
1112 |
1029 |
0.14 |
|
|
|
| 21 |
A' |
1019 |
943 |
0.89 |
|
|
|
| 22 |
A' |
1009 |
934 |
0.67 |
|
|
|
| 23 |
A' |
916 |
848 |
0.92 |
|
|
|
| 24 |
A' |
851 |
788 |
0.96 |
|
|
|
| 25 |
A' |
682 |
632 |
1.61 |
|
|
|
| 26 |
A' |
457 |
424 |
0.06 |
|
|
|
| 27 |
A' |
344 |
319 |
0.10 |
|
|
|
| 28 |
A' |
117 |
108 |
0.01 |
|
|
|
| 29 |
A" |
3229 |
2989 |
38.97 |
|
|
|
| 30 |
A" |
3225 |
2986 |
30.12 |
|
|
|
| 31 |
A" |
3220 |
2981 |
9.24 |
|
|
|
| 32 |
A" |
3167 |
2932 |
64.58 |
|
|
|
| 33 |
A" |
1597 |
1478 |
2.39 |
|
|
|
| 34 |
A" |
1594 |
1476 |
0.42 |
|
|
|
| 35 |
A" |
1577 |
1460 |
3.62 |
|
|
|
| 36 |
A" |
1388 |
1285 |
0.61 |
|
|
|
| 37 |
A" |
1358 |
1258 |
0.00 |
|
|
|
| 38 |
A" |
1320 |
1222 |
0.08 |
|
|
|
| 39 |
A" |
1212 |
1123 |
0.52 |
|
|
|
| 40 |
A" |
1203 |
1114 |
0.09 |
|
|
|
| 41 |
A" |
1054 |
976 |
2.81 |
|
|
|
| 42 |
A" |
966 |
895 |
0.02 |
|
|
|
| 43 |
A" |
961 |
890 |
0.10 |
|
|
|
| 44 |
A" |
860 |
796 |
0.21 |
|
|
|
| 45 |
A" |
392 |
363 |
0.01 |
|
|
|
| 46 |
A" |
272 |
252 |
0.11 |
|
|
|
| 47 |
A" |
260 |
240 |
0.00 |
|
|
|
| 48 |
A" |
243 |
225 |
0.02 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 38526.4 cm
-1
Scaled (by 0.9258) Zero Point Vibrational Energy (zpe) 35667.7 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at CISD/6-31G*
Point Group is Cs
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.272 |
0.096 |
1.074 |
| C2 |
0.272 |
0.096 |
-1.074 |
| C3 |
-0.664 |
0.691 |
0.000 |
| C4 |
0.869 |
-0.842 |
0.000 |
| C5 |
-0.917 |
2.184 |
0.000 |
| C6 |
0.272 |
-2.239 |
0.000 |
| H7 |
0.998 |
0.833 |
1.417 |
| H8 |
-0.186 |
-0.369 |
1.946 |
| H9 |
0.998 |
0.833 |
-1.417 |
| H10 |
-0.186 |
-0.369 |
-1.946 |
| H11 |
-1.616 |
0.159 |
0.000 |
| H12 |
1.956 |
-0.912 |
0.000 |
| H13 |
0.024 |
2.735 |
0.000 |
| H14 |
-0.818 |
-2.205 |
0.000 |
| H15 |
-1.482 |
2.488 |
-0.881 |
| H16 |
-1.482 |
2.488 |
0.881 |
| H17 |
0.586 |
-2.797 |
-0.882 |
| H18 |
0.586 |
-2.797 |
0.882 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
C3 |
C4 |
C5 |
C6 |
H7 |
H8 |
H9 |
H10 |
H11 |
H12 |
H13 |
H14 |
H15 |
H16 |
H17 |
H18 |
| C1 | | 2.1474 | 1.5435 | 1.5452 | 2.6321 | 2.5700 | 1.0901 | 1.0897 | 2.6972 | 3.0897 | 2.1726 | 2.2374 | 2.8598 | 2.7629 | 3.5530 | 2.9731 | 3.5062 | 2.9166 |
C2 | 2.1474 | | 1.5435 | 1.5452 | 2.6321 | 2.5700 | 2.6972 | 3.0897 | 1.0901 | 1.0897 | 2.1726 | 2.2374 | 2.8598 | 2.7629 | 2.9731 | 3.5530 | 2.9166 | 3.5062 | C3 | 1.5435 | 1.5435 | | 2.1671 | 1.5148 | 3.0755 | 2.1890 | 2.2669 | 2.1890 | 2.2669 | 1.0904 | 3.0715 | 2.1567 | 2.8996 | 2.1630 | 2.1630 | 3.8087 | 3.8087 | C4 | 1.5452 | 1.5452 | 2.1671 | | 3.5135 | 1.5196 | 2.1972 | 2.2637 | 2.1972 | 2.2637 | 2.6783 | 1.0897 | 3.6749 | 2.1685 | 4.1706 | 4.1706 | 2.1639 | 2.1639 | C5 | 2.6321 | 2.6321 | 1.5148 | 3.5135 | | 4.5803 | 2.7396 | 3.2925 | 2.7396 | 3.2925 | 2.1423 | 4.2244 | 1.0903 | 4.3901 | 1.0901 | 1.0901 | 5.2776 | 5.2776 | C6 | 2.5700 | 2.5700 | 3.0755 | 1.5196 | 4.5803 | | 3.4599 | 2.7377 | 3.4599 | 2.7377 | 3.0517 | 2.1440 | 4.9801 | 1.0901 | 5.1189 | 5.1189 | 1.0899 | 1.0899 | H7 | 1.0901 | 2.6972 | 2.1890 | 2.1972 | 2.7396 | 3.4599 | | 1.7683 | 2.8345 | 3.7630 | 3.0489 | 2.4435 | 2.5647 | 3.8121 | 3.7656 | 3.0305 | 4.3164 | 3.6923 | H8 | 1.0897 | 3.0897 | 2.2669 | 2.2637 | 3.2925 | 2.7377 | 1.7683 | | 3.7630 | 3.8925 | 2.4718 | 2.9450 | 3.6696 | 2.7490 | 4.2237 | 3.3135 | 3.8067 | 2.7617 | H9 | 2.6972 | 1.0901 | 2.1890 | 2.1972 | 2.7396 | 3.4599 | 2.8345 | 3.7630 | | 1.7683 | 3.0489 | 2.4435 | 2.5647 | 3.8121 | 3.0305 | 3.7656 | 3.6923 | 4.3164 | H10 | 3.0897 | 1.0897 | 2.2669 | 2.2637 | 3.2925 | 2.7377 | 3.7630 | 3.8925 | 1.7683 | | 2.4718 | 2.9450 | 3.6696 | 2.7490 | 3.3135 | 4.2237 | 2.7617 | 3.8067 | H11 | 2.1726 | 2.1726 | 1.0904 | 2.6783 | 2.1423 | 3.0517 | 3.0489 | 2.4718 | 3.0489 | 2.4718 | | 3.7290 | 3.0534 | 2.4947 | 2.4943 | 2.4943 | 3.7901 | 3.7901 | H12 | 2.2374 | 2.2374 | 3.0715 | 1.0897 | 4.2244 | 2.1440 | 2.4435 | 2.9450 | 2.4435 | 2.9450 | 3.7290 | | 4.1276 | 3.0601 | 4.9159 | 4.9159 | 2.4915 | 2.4915 | H13 | 2.8598 | 2.8598 | 2.1567 | 3.6749 | 1.0903 | 4.9801 | 2.5647 | 3.6696 | 2.5647 | 3.6696 | 3.0534 | 4.1276 | | 5.0108 | 1.7622 | 1.7622 | 5.6302 | 5.6302 | H14 | 2.7629 | 2.7629 | 2.8996 | 2.1685 | 4.3901 | 1.0901 | 3.8121 | 2.7490 | 3.8121 | 2.7490 | 2.4947 | 3.0601 | 5.0108 | | 4.8213 | 4.8213 | 1.7604 | 1.7604 | H15 | 3.5530 | 2.9731 | 2.1630 | 4.1706 | 1.0901 | 5.1189 | 3.7656 | 4.2237 | 3.0305 | 3.3135 | 2.4943 | 4.9159 | 1.7622 | 4.8213 | | 1.7623 | 5.6762 | 5.9437 | H16 | 2.9731 | 3.5530 | 2.1630 | 4.1706 | 1.0901 | 5.1189 | 3.0305 | 3.3135 | 3.7656 | 4.2237 | 2.4943 | 4.9159 | 1.7622 | 4.8213 | 1.7623 | | 5.9437 | 5.6762 | H17 | 3.5062 | 2.9166 | 3.8087 | 2.1639 | 5.2776 | 1.0899 | 4.3164 | 3.8067 | 3.6923 | 2.7617 | 3.7901 | 2.4915 | 5.6302 | 1.7604 | 5.6762 | 5.9437 | | 1.7633 | H18 | 2.9166 | 3.5062 | 3.8087 | 2.1639 | 5.2776 | 1.0899 | 3.6923 | 2.7617 | 4.3164 | 3.8067 | 3.7901 | 2.4915 | 5.6302 | 1.7604 | 5.9437 | 5.6762 | 1.7633 | |
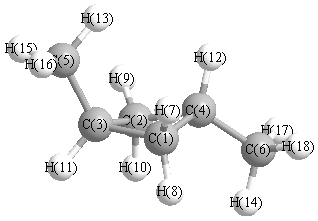 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C3 |
C2 |
88.154 |
|
C1 |
C3 |
C5 |
118.776 |
| C1 |
C3 |
H11 |
109.959 |
|
C1 |
C4 |
C2 |
88.029 |
| C1 |
C4 |
C6 |
113.968 |
|
C1 |
C4 |
H12 |
115.140 |
| C2 |
C3 |
C5 |
118.776 |
|
C2 |
C3 |
H11 |
109.959 |
| C2 |
C4 |
C6 |
113.968 |
|
C2 |
C4 |
H12 |
115.140 |
| C3 |
C1 |
C4 |
89.117 |
|
C3 |
C1 |
H7 |
111.277 |
| C3 |
C1 |
H8 |
117.802 |
|
C3 |
C2 |
C4 |
89.117 |
| C3 |
C2 |
H9 |
111.277 |
|
C3 |
C2 |
H10 |
117.802 |
| C3 |
C5 |
H13 |
110.704 |
|
C3 |
C5 |
H15 |
111.224 |
| C3 |
C5 |
H16 |
111.224 |
|
C4 |
C1 |
H7 |
111.814 |
| C4 |
C1 |
H8 |
117.387 |
|
C4 |
C2 |
H9 |
111.814 |
| C4 |
C2 |
H10 |
117.387 |
|
C4 |
C6 |
H14 |
111.327 |
| C4 |
C6 |
H17 |
110.971 |
|
C4 |
C6 |
H18 |
110.971 |
| C5 |
C3 |
H11 |
109.559 |
|
C6 |
C4 |
H12 |
109.399 |
| H7 |
C1 |
H8 |
108.428 |
|
H9 |
C2 |
H10 |
108.428 |
| H13 |
C5 |
H15 |
107.838 |
|
H13 |
C5 |
H16 |
107.838 |
| H14 |
C6 |
H17 |
107.714 |
|
H14 |
C6 |
H18 |
107.714 |
| H15 |
C5 |
H16 |
107.864 |
|
H17 |
C6 |
H18 |
107.987 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability