Jump to
S1C2
S1C3
Vibrational Frequencies calculated at CISD/6-311G*
Geometric Data calculated at CISD/6-311G*
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Jump to
S1C1
S1C3
Energy calculated at CISD/6-311G*
| | hartrees |
|---|
| Energy at 0K | -351.002379 |
| Energy at 298.15K | -351.003680 |
| HF Energy | -350.604783 |
| Nuclear repulsion energy | 82.574585 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at CISD/6-311G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
4171 |
3860 |
2.27 |
|
|
|
| 2 |
A |
633 |
586 |
5.55 |
|
|
|
| 3 |
A |
247 |
229 |
83.28 |
|
|
|
| 4 |
A |
160 |
148 |
41.74 |
|
|
|
| 5 |
A |
103 |
95 |
271.09 |
|
|
|
| 6 |
B |
4169 |
3858 |
101.81 |
|
|
|
| 7 |
B |
947 |
876 |
140.29 |
|
|
|
| 8 |
B |
243 |
225 |
345.62 |
|
|
|
| 9 |
B |
154 |
142 |
30.33 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 5413.9 cm
-1
Scaled (by 0.9253) Zero Point Vibrational Energy (zpe) 5009.4 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at CISD/6-311G*
Point Group is C2
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| Mg1 |
0.000 |
0.000 |
0.003 |
| O2 |
0.000 |
1.775 |
-0.027 |
| O3 |
0.000 |
-1.775 |
-0.027 |
| H4 |
0.343 |
2.623 |
0.196 |
| H5 |
-0.343 |
-2.623 |
0.196 |
Atom - Atom Distances (Å)
| |
Mg1 |
O2 |
O3 |
H4 |
H5 |
| Mg1 | | 1.7753 | 1.7753 | 2.6520 | 2.6520 |
O2 | 1.7753 | | 3.5501 | 0.9411 | 4.4167 | O3 | 1.7753 | 3.5501 | | 4.4167 | 0.9411 | H4 | 2.6520 | 0.9411 | 4.4167 | | 5.2900 | H5 | 2.6520 | 4.4167 | 0.9411 | 5.2900 | |
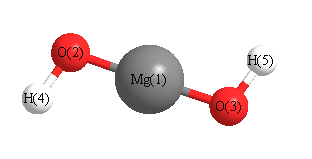 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| Mg1 |
O2 |
H4 |
153.715 |
|
Mg1 |
O3 |
H5 |
153.715 |
| O2 |
Mg1 |
O3 |
178.060 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Jump to
S1C1
S1C2
Energy calculated at CISD/6-311G*
| | hartrees |
|---|
| Energy at 0K | -351.001978 |
| Energy at 298.15K | |
| HF Energy | -350.605691 |
| Nuclear repulsion energy | 83.069261 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at CISD/6-311G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
Σg |
4202 |
3888 |
0.00 |
|
|
|
| 2 |
Σg |
635 |
588 |
0.00 |
|
|
|
| 3 |
Σu |
4199 |
3885 |
136.07 |
|
|
|
| 4 |
Σu |
961 |
889 |
174.52 |
|
|
|
| 5 |
Πg |
213i |
197i |
0.00 |
|
|
|
| 5 |
Πg |
213i |
197i |
0.00 |
|
|
|
| 6 |
Πu |
157 |
146 |
25.11 |
|
|
|
| 6 |
Πu |
157 |
146 |
25.11 |
|
|
|
| 7 |
Πu |
161i |
149i |
407.13 |
|
|
|
| 7 |
Πu |
161i |
149i |
407.13 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 4782.4 cm
-1
Scaled (by 0.9253) Zero Point Vibrational Energy (zpe) 4425.2 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at CISD/6-311G*
Point Group is D∞h
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| Mg1 |
0.000 |
0.000 |
0.000 |
| O2 |
0.000 |
0.000 |
1.760 |
| O3 |
0.000 |
0.000 |
-1.760 |
| H4 |
0.000 |
0.000 |
2.699 |
| H5 |
0.000 |
0.000 |
-2.699 |
Atom - Atom Distances (Å)
| |
Mg1 |
O2 |
O3 |
H4 |
H5 |
| Mg1 | | 1.7600 | 1.7600 | 2.6991 | 2.6991 |
O2 | 1.7600 | | 3.5199 | 0.9391 | 4.4591 | O3 | 1.7600 | 3.5199 | | 4.4591 | 0.9391 | H4 | 2.6991 | 0.9391 | 4.4591 | | 5.3982 | H5 | 2.6991 | 4.4591 | 0.9391 | 5.3982 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| Mg1 |
O2 |
H4 |
180.000 |
|
Mg1 |
O3 |
H5 |
180.000 |
| O2 |
Mg1 |
O3 |
180.000 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability