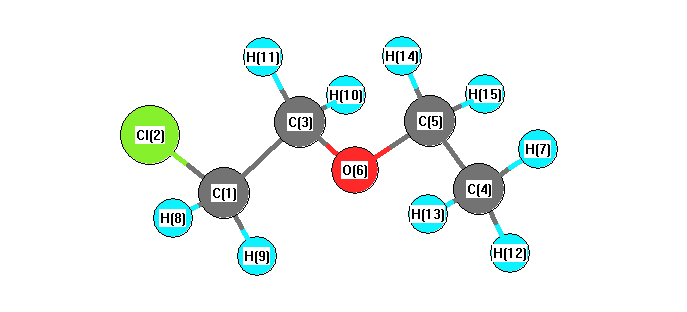
Vibrational Frequencies calculated at B3PW91/6-31G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3151 |
3014 |
23.90 |
|
|
|
| 2 |
A' |
3123 |
2988 |
15.35 |
|
|
|
| 3 |
A' |
3070 |
2937 |
16.48 |
|
|
|
| 4 |
A' |
3003 |
2873 |
55.63 |
|
|
|
| 5 |
A' |
2986 |
2856 |
32.90 |
|
|
|
| 6 |
A' |
1562 |
1494 |
1.67 |
|
|
|
| 7 |
A' |
1541 |
1475 |
5.68 |
|
|
|
| 8 |
A' |
1526 |
1460 |
4.69 |
|
|
|
| 9 |
A' |
1516 |
1450 |
3.43 |
|
|
|
| 10 |
A' |
1467 |
1404 |
13.00 |
|
|
|
| 11 |
A' |
1428 |
1366 |
15.36 |
|
|
|
| 12 |
A' |
1400 |
1340 |
27.62 |
|
|
|
| 13 |
A' |
1303 |
1247 |
24.56 |
|
|
|
| 14 |
A' |
1192 |
1140 |
223.70 |
|
|
|
| 15 |
A' |
1166 |
1115 |
36.72 |
|
|
|
| 16 |
A' |
1087 |
1040 |
1.94 |
|
|
|
| 17 |
A' |
1062 |
1016 |
14.80 |
|
|
|
| 18 |
A' |
916 |
876 |
11.72 |
|
|
|
| 19 |
A' |
777 |
743 |
46.95 |
|
|
|
| 20 |
A' |
480 |
459 |
0.53 |
|
|
|
| 21 |
A' |
378 |
362 |
2.67 |
|
|
|
| 22 |
A' |
267 |
255 |
3.48 |
|
|
|
| 23 |
A' |
122 |
117 |
1.40 |
|
|
|
| 24 |
A" |
3189 |
3051 |
7.72 |
|
|
|
| 25 |
A" |
3155 |
3018 |
25.35 |
|
|
|
| 26 |
A" |
3039 |
2907 |
37.42 |
|
|
|
| 27 |
A" |
3018 |
2887 |
70.05 |
|
|
|
| 28 |
A" |
1509 |
1444 |
6.21 |
|
|
|
| 29 |
A" |
1314 |
1257 |
2.04 |
|
|
|
| 30 |
A" |
1310 |
1253 |
0.55 |
|
|
|
| 31 |
A" |
1233 |
1179 |
5.34 |
|
|
|
| 32 |
A" |
1192 |
1140 |
4.11 |
|
|
|
| 33 |
A" |
1080 |
1033 |
2.59 |
|
|
|
| 34 |
A" |
830 |
794 |
1.16 |
|
|
|
| 35 |
A" |
810 |
775 |
0.42 |
|
|
|
| 36 |
A" |
256 |
244 |
0.90 |
|
|
|
| 37 |
A" |
154 |
147 |
7.83 |
|
|
|
| 38 |
A" |
86 |
82 |
2.18 |
|
|
|
| 39 |
A" |
58 |
56 |
0.32 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 28375.4 cm
-1
Scaled (by 0.9567) Zero Point Vibrational Energy (zpe) 27146.7 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B3PW91/6-31G*
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.441 |
|
|
-0.205 |
| 2 |
Cl |
-0.074 |
|
|
-0.173 |
| 3 |
C |
-0.044 |
|
|
0.160 |
| 4 |
C |
-0.524 |
|
|
-0.316 |
| 5 |
C |
-0.056 |
|
|
0.295 |
| 6 |
O |
-0.474 |
|
|
-0.382 |
| 7 |
H |
0.170 |
|
|
0.081 |
| 8 |
H |
0.228 |
|
|
0.152 |
| 9 |
H |
0.228 |
|
|
0.152 |
| 10 |
H |
0.163 |
|
|
0.035 |
| 11 |
H |
0.163 |
|
|
0.035 |
| 12 |
H |
0.184 |
|
|
0.094 |
| 13 |
H |
0.184 |
|
|
0.094 |
| 14 |
H |
0.147 |
|
|
-0.011 |
| 15 |
H |
0.147 |
|
|
-0.011 |
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
2.145 |
-0.314 |
0.000 |
2.167 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
2.165 |
-0.383 |
0.000 |
2.199 |
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-45.911 |
3.213 |
0.000 |
| y |
3.213 |
-45.410 |
0.000 |
| z |
0.000 |
0.000 |
-43.834 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-1.289 |
3.213 |
0.000 |
| y |
3.213 |
-0.537 |
0.000 |
| z |
0.000 |
0.000 |
1.826 |
|
| Polar |
|---|
| 3z2-r2 | 3.653 |
|---|
| x2-y2 | -0.501 |
|---|
| xy | 3.213 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
0.000 |
0.000 |
0.000 |
| y |
0.000 |
0.000 |
0.000 |
| z |
0.000 |
0.000 |
0.000 |
<r2> (average value of r
2) Å
2
| <r2> |
365.351 |
| (<r2>)1/2 |
19.114 |