Vibrational Frequencies calculated at B3PW91/6-31G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3133 |
2998 |
34.28 |
|
|
|
| 2 |
A |
3114 |
2980 |
12.72 |
|
|
|
| 3 |
A |
3065 |
2932 |
29.38 |
|
|
|
| 4 |
A |
3052 |
2919 |
28.26 |
|
|
|
| 5 |
A |
3045 |
2913 |
39.34 |
|
|
|
| 6 |
A |
3028 |
2897 |
23.47 |
|
|
|
| 7 |
A |
2378 |
2275 |
0.02 |
|
|
|
| 8 |
A |
1532 |
1466 |
5.20 |
|
|
|
| 9 |
A |
1517 |
1452 |
0.64 |
|
|
|
| 10 |
A |
1504 |
1439 |
6.78 |
|
|
|
| 11 |
A |
1499 |
1434 |
1.29 |
|
|
|
| 12 |
A |
1438 |
1375 |
3.87 |
|
|
|
| 13 |
A |
1437 |
1375 |
2.53 |
|
|
|
| 14 |
A |
1401 |
1341 |
2.80 |
|
|
|
| 15 |
A |
1317 |
1260 |
12.65 |
|
|
|
| 16 |
A |
1186 |
1135 |
0.29 |
|
|
|
| 17 |
A |
1123 |
1075 |
3.60 |
|
|
|
| 18 |
A |
1063 |
1017 |
1.47 |
|
|
|
| 19 |
A |
1062 |
1016 |
0.88 |
|
|
|
| 20 |
A |
908 |
868 |
3.46 |
|
|
|
| 21 |
A |
778 |
745 |
0.27 |
|
|
|
| 22 |
A |
487 |
466 |
1.27 |
|
|
|
| 23 |
A |
338 |
323 |
0.27 |
|
|
|
| 24 |
A |
277 |
265 |
5.66 |
|
|
|
| 25 |
A |
102 |
98 |
1.47 |
|
|
|
| 26 |
A |
3128 |
2993 |
61.35 |
|
|
|
| 27 |
A |
3114 |
2980 |
12.66 |
|
|
|
| 28 |
A |
3102 |
2968 |
4.34 |
|
|
|
| 29 |
A |
3061 |
2928 |
9.53 |
|
|
|
| 30 |
A |
1525 |
1459 |
7.88 |
|
|
|
| 31 |
A |
1504 |
1439 |
6.37 |
|
|
|
| 32 |
A |
1335 |
1278 |
0.02 |
|
|
|
| 33 |
A |
1270 |
1215 |
0.14 |
|
|
|
| 34 |
A |
1132 |
1083 |
0.60 |
|
|
|
| 35 |
A |
1062 |
1016 |
1.77 |
|
|
|
| 36 |
A |
880 |
842 |
0.56 |
|
|
|
| 37 |
A |
748 |
716 |
3.49 |
|
|
|
| 38 |
A |
377 |
361 |
0.00 |
|
|
|
| 39 |
A |
246 |
235 |
0.10 |
|
|
|
| 40 |
A |
221 |
212 |
8.00 |
|
|
|
| 41 |
A |
84 |
81 |
0.31 |
|
|
|
| 42 |
A |
18 |
18 |
0.00 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 31296.6 cm
-1
Scaled (by 0.9567) Zero Point Vibrational Energy (zpe) 29941.5 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
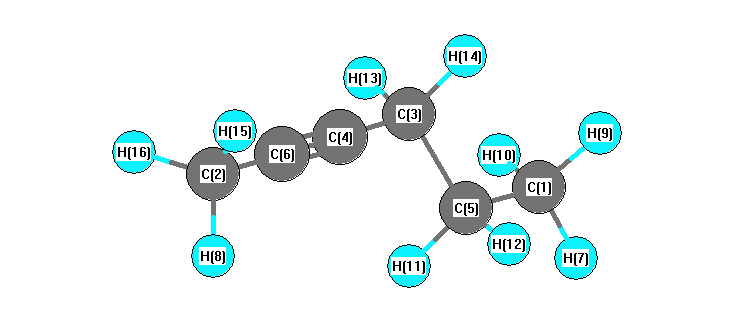
Charges from optimized geometry at B3PW91/6-31G*
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.510 |
|
|
-0.430 |
| 2 |
C |
-0.636 |
|
|
-0.334 |
| 3 |
C |
-0.441 |
|
|
0.167 |
| 4 |
C |
0.093 |
|
|
-0.352 |
| 5 |
C |
-0.306 |
|
|
0.092 |
| 6 |
C |
-0.025 |
|
|
0.056 |
| 7 |
H |
0.172 |
|
|
0.120 |
| 8 |
H |
0.200 |
|
|
0.122 |
| 9 |
H |
0.166 |
|
|
0.099 |
| 10 |
H |
0.166 |
|
|
0.099 |
| 11 |
H |
0.173 |
|
|
0.018 |
| 12 |
H |
0.173 |
|
|
0.018 |
| 13 |
H |
0.187 |
|
|
0.032 |
| 14 |
H |
0.187 |
|
|
0.032 |
| 15 |
H |
0.200 |
|
|
0.130 |
| 16 |
H |
0.200 |
|
|
0.130 |
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
-0.120 |
-0.086 |
0.000 |
0.148 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
-0.121 |
-0.057 |
0.000 |
0.134 |
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-35.620 |
-3.025 |
0.000 |
| y |
-3.025 |
-34.324 |
0.000 |
| z |
0.000 |
0.000 |
-37.843 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
0.463 |
-3.025 |
0.000 |
| y |
-3.025 |
2.408 |
0.000 |
| z |
0.000 |
0.000 |
-2.871 |
|
| Polar |
|---|
| 3z2-r2 | -5.741 |
|---|
| x2-y2 | -1.296 |
|---|
| xy | -3.025 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
0.000 |
0.000 |
0.000 |
| y |
0.000 |
0.000 |
0.000 |
| z |
0.000 |
0.000 |
0.000 |
<r2> (average value of r
2) Å
2
| <r2> |
293.911 |
| (<r2>)1/2 |
17.144 |