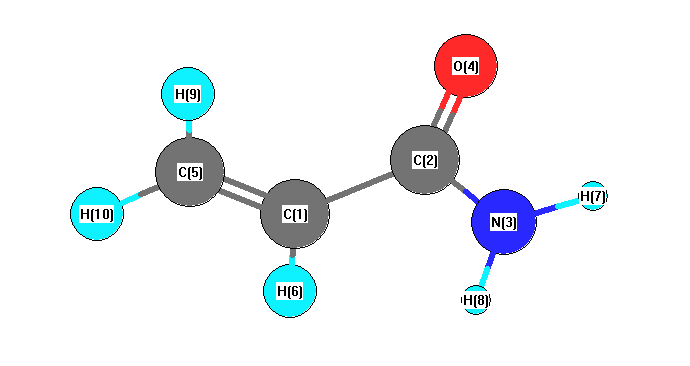
Vibrational Frequencies calculated at B3PW91/6-31G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3748 |
3585 |
37.46 |
|
|
|
| 2 |
A |
3618 |
3461 |
45.43 |
|
|
|
| 3 |
A |
3278 |
3136 |
2.90 |
|
|
|
| 4 |
A |
3188 |
3050 |
10.01 |
|
|
|
| 5 |
A |
3174 |
3036 |
12.59 |
|
|
|
| 6 |
A |
1815 |
1736 |
232.96 |
|
|
|
| 7 |
A |
1719 |
1645 |
30.71 |
|
|
|
| 8 |
A |
1647 |
1576 |
127.60 |
|
|
|
| 9 |
A |
1454 |
1391 |
94.47 |
|
|
|
| 10 |
A |
1374 |
1315 |
51.61 |
|
|
|
| 11 |
A |
1302 |
1245 |
75.76 |
|
|
|
| 12 |
A |
1124 |
1075 |
2.44 |
|
|
|
| 13 |
A |
1043 |
998 |
3.69 |
|
|
|
| 14 |
A |
1021 |
977 |
36.10 |
|
|
|
| 15 |
A |
989 |
947 |
12.48 |
|
|
|
| 16 |
A |
826 |
791 |
6.63 |
|
|
|
| 17 |
A |
818 |
783 |
27.16 |
|
|
|
| 18 |
A |
624 |
597 |
12.70 |
|
|
|
| 19 |
A |
612 |
585 |
6.53 |
|
|
|
| 20 |
A |
470 |
450 |
9.48 |
|
|
|
| 21 |
A |
464 |
444 |
7.18 |
|
|
|
| 22 |
A |
275 |
263 |
7.37 |
|
|
|
| 23 |
A |
171 |
164 |
199.57 |
|
|
|
| 24 |
A |
109 |
104 |
61.49 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 17431.5 cm
-1
Scaled (by 0.9567) Zero Point Vibrational Energy (zpe) 16676.7 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B3PW91/6-31G*
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.176 |
|
|
-0.305 |
| 2 |
C |
0.592 |
|
|
0.739 |
| 3 |
N |
-0.799 |
|
|
-0.954 |
| 4 |
O |
-0.518 |
|
|
-0.524 |
| 5 |
C |
-0.354 |
|
|
-0.262 |
| 6 |
H |
0.157 |
|
|
0.160 |
| 7 |
H |
0.362 |
|
|
0.419 |
| 8 |
H |
0.354 |
|
|
0.405 |
| 9 |
H |
0.208 |
|
|
0.161 |
| 10 |
H |
0.176 |
|
|
0.160 |
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
-0.455 |
-3.499 |
0.319 |
3.543 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
-0.440 |
-3.504 |
0.379 |
3.552 |
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-21.275 |
2.955 |
-0.648 |
| y |
2.955 |
-29.480 |
-0.319 |
| z |
-0.648 |
-0.319 |
-31.307 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
9.119 |
2.955 |
-0.648 |
| y |
2.955 |
-3.189 |
-0.319 |
| z |
-0.648 |
-0.319 |
-5.929 |
|
| Polar |
|---|
| 3z2-r2 | -11.858 |
|---|
| x2-y2 | 8.205 |
|---|
| xy | 2.955 |
|---|
| xz | -0.648 |
|---|
| yz | -0.319 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
0.000 |
0.000 |
0.000 |
| y |
0.000 |
0.000 |
0.000 |
| z |
0.000 |
0.000 |
0.000 |
<r2> (average value of r
2) Å
2
| <r2> |
114.928 |
| (<r2>)1/2 |
10.720 |