Vibrational Frequencies calculated at B3PW91/aug-cc-pVTZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3206 |
3094 |
7.91 |
|
|
|
| 2 |
A' |
3203 |
3091 |
6.52 |
|
|
|
| 3 |
A' |
3193 |
3081 |
5.16 |
|
|
|
| 4 |
A' |
3186 |
3075 |
4.56 |
|
|
|
| 5 |
A' |
3175 |
3064 |
0.47 |
|
|
|
| 6 |
A' |
1661 |
1603 |
23.00 |
|
|
|
| 7 |
A' |
1646 |
1588 |
2.44 |
|
|
|
| 8 |
A' |
1596 |
1541 |
188.48 |
|
|
|
| 9 |
A' |
1501 |
1449 |
8.69 |
|
|
|
| 10 |
A' |
1487 |
1435 |
32.65 |
|
|
|
| 11 |
A' |
1377 |
1329 |
14.71 |
|
|
|
| 12 |
A' |
1333 |
1287 |
7.49 |
|
|
|
| 13 |
A' |
1197 |
1155 |
23.27 |
|
|
|
| 14 |
A' |
1179 |
1138 |
1.49 |
|
|
|
| 15 |
A' |
1131 |
1091 |
145.12 |
|
|
|
| 16 |
A' |
1096 |
1057 |
5.46 |
|
|
|
| 17 |
A' |
1039 |
1003 |
6.84 |
|
|
|
| 18 |
A' |
1020 |
984 |
1.51 |
|
|
|
| 19 |
A' |
835 |
806 |
39.26 |
|
|
|
| 20 |
A' |
682 |
658 |
9.35 |
|
|
|
| 21 |
A' |
620 |
598 |
0.11 |
|
|
|
| 22 |
A' |
447 |
431 |
1.19 |
|
|
|
| 23 |
A' |
255 |
246 |
1.89 |
|
|
|
| 24 |
A" |
1014 |
979 |
0.04 |
|
|
|
| 25 |
A" |
1001 |
966 |
0.01 |
|
|
|
| 26 |
A" |
963 |
930 |
4.00 |
|
|
|
| 27 |
A" |
865 |
835 |
0.01 |
|
|
|
| 28 |
A" |
777 |
750 |
62.48 |
|
|
|
| 29 |
A" |
683 |
659 |
20.58 |
|
|
|
| 30 |
A" |
466 |
450 |
2.12 |
|
|
|
| 31 |
A" |
413 |
399 |
0.00 |
|
|
|
| 32 |
A" |
243 |
235 |
0.34 |
|
|
|
| 33 |
A" |
107 |
103 |
0.09 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 21298.8 cm
-1
Scaled (by 0.9651) Zero Point Vibrational Energy (zpe) 20555.5 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
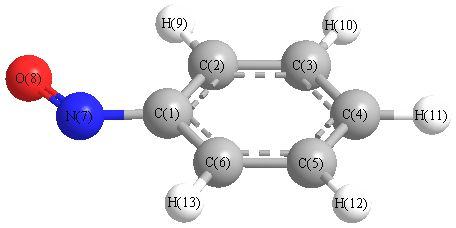
Charges from optimized geometry at B3PW91/aug-cc-pVTZ
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
1.079 |
|
|
|
| 2 |
C |
-0.704 |
|
|
|
| 3 |
C |
-0.349 |
|
|
|
| 4 |
C |
-0.411 |
|
|
|
| 5 |
C |
-0.423 |
|
|
|
| 6 |
C |
-0.869 |
|
|
|
| 7 |
N |
-0.343 |
|
|
|
| 8 |
O |
-0.434 |
|
|
|
| 9 |
H |
0.538 |
|
|
|
| 10 |
H |
0.430 |
|
|
|
| 11 |
H |
0.519 |
|
|
|
| 12 |
H |
0.376 |
|
|
|
| 13 |
H |
0.591 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
1.125 |
-3.635 |
0.000 |
3.806 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-41.726 |
2.336 |
0.000 |
| y |
2.336 |
-48.764 |
0.000 |
| z |
0.000 |
0.000 |
-47.579 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
6.446 |
2.336 |
0.000 |
| y |
2.336 |
-4.111 |
0.000 |
| z |
0.000 |
0.000 |
-2.334 |
|
| Polar |
|---|
| 3z2-r2 | -4.669 |
|---|
| x2-y2 | 7.038 |
|---|
| xy | 2.336 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
13.664 |
-1.710 |
0.000 |
| y |
-1.710 |
16.737 |
0.000 |
| z |
0.000 |
0.000 |
7.020 |
<r2> (average value of r
2) Å
2
| <r2> |
247.942 |
| (<r2>)1/2 |
15.746 |