Vibrational Frequencies calculated at B3PW91/6-31G(2df,p)
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3711 |
3568 |
70.20 |
|
|
|
| 2 |
A' |
3276 |
3149 |
3.63 |
|
|
|
| 3 |
A' |
3256 |
3130 |
0.72 |
|
|
|
| 4 |
A' |
3208 |
3084 |
17.66 |
|
|
|
| 5 |
A' |
3196 |
3073 |
29.63 |
|
|
|
| 6 |
A' |
3185 |
3062 |
2.81 |
|
|
|
| 7 |
A' |
3179 |
3056 |
1.38 |
|
|
|
| 8 |
A' |
1674 |
1609 |
2.87 |
|
|
|
| 9 |
A' |
1629 |
1567 |
1.95 |
|
|
|
| 10 |
A' |
1563 |
1503 |
6.50 |
|
|
|
| 11 |
A' |
1530 |
1471 |
4.25 |
|
|
|
| 12 |
A' |
1489 |
1431 |
22.78 |
|
|
|
| 13 |
A' |
1458 |
1402 |
11.84 |
|
|
|
| 14 |
A' |
1387 |
1333 |
34.99 |
|
|
|
| 15 |
A' |
1379 |
1326 |
4.82 |
|
|
|
| 16 |
A' |
1312 |
1261 |
8.66 |
|
|
|
| 17 |
A' |
1266 |
1217 |
10.06 |
|
|
|
| 18 |
A' |
1227 |
1180 |
3.57 |
|
|
|
| 19 |
A' |
1171 |
1126 |
1.01 |
|
|
|
| 20 |
A' |
1145 |
1101 |
1.15 |
|
|
|
| 21 |
A' |
1114 |
1071 |
21.88 |
|
|
|
| 22 |
A' |
1092 |
1050 |
4.05 |
|
|
|
| 23 |
A' |
1040 |
1000 |
4.57 |
|
|
|
| 24 |
A' |
908 |
873 |
6.08 |
|
|
|
| 25 |
A' |
881 |
847 |
0.32 |
|
|
|
| 26 |
A' |
776 |
746 |
2.60 |
|
|
|
| 27 |
A' |
613 |
589 |
1.09 |
|
|
|
| 28 |
A' |
548 |
527 |
0.06 |
|
|
|
| 29 |
A' |
402 |
386 |
3.70 |
|
|
|
| 30 |
A" |
963 |
926 |
0.02 |
|
|
|
| 31 |
A" |
919 |
884 |
1.49 |
|
|
|
| 32 |
A" |
864 |
831 |
1.05 |
|
|
|
| 33 |
A" |
848 |
815 |
2.47 |
|
|
|
| 34 |
A" |
788 |
758 |
9.77 |
|
|
|
| 35 |
A" |
759 |
730 |
65.58 |
|
|
|
| 36 |
A" |
736 |
708 |
28.72 |
|
|
|
| 37 |
A" |
619 |
595 |
5.30 |
|
|
|
| 38 |
A" |
586 |
563 |
0.70 |
|
|
|
| 39 |
A" |
434 |
417 |
0.12 |
|
|
|
| 40 |
A" |
408 |
393 |
72.25 |
|
|
|
| 41 |
A" |
246 |
237 |
0.04 |
|
|
|
| 42 |
A" |
218 |
209 |
8.93 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 28500.2 cm
-1
Scaled (by 0.9614) Zero Point Vibrational Energy (zpe) 27400.1 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
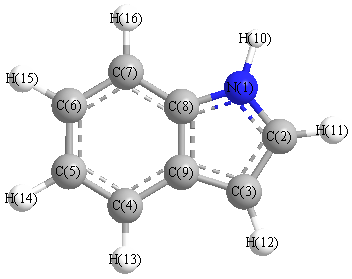
Charges from optimized geometry at B3PW91/6-31G(2df,p)
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
N |
-0.422 |
|
|
|
| 2 |
C |
-0.064 |
|
|
|
| 3 |
C |
-0.324 |
|
|
|
| 4 |
C |
-0.268 |
|
|
|
| 5 |
C |
-0.146 |
|
|
|
| 6 |
C |
-0.140 |
|
|
|
| 7 |
C |
-0.278 |
|
|
|
| 8 |
C |
0.339 |
|
|
|
| 9 |
C |
0.275 |
|
|
|
| 10 |
H |
0.282 |
|
|
|
| 11 |
H |
0.149 |
|
|
|
| 12 |
H |
0.127 |
|
|
|
| 13 |
H |
0.119 |
|
|
|
| 14 |
H |
0.117 |
|
|
|
| 15 |
H |
0.119 |
|
|
|
| 16 |
H |
0.114 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
-0.914 |
1.984 |
0.000 |
2.184 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-45.839 |
-2.133 |
0.000 |
| y |
-2.133 |
-41.277 |
0.000 |
| z |
0.000 |
0.000 |
-55.890 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
2.744 |
-2.133 |
0.000 |
| y |
-2.133 |
9.588 |
0.000 |
| z |
0.000 |
0.000 |
-12.332 |
|
| Polar |
|---|
| 3z2-r2 | -24.664 |
|---|
| x2-y2 | -4.563 |
|---|
| xy | -2.133 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
18.144 |
-1.477 |
0.000 |
| y |
-1.477 |
14.648 |
0.000 |
| z |
0.000 |
0.000 |
5.397 |
<r2> (average value of r
2) Å
2
| <r2> |
280.280 |
| (<r2>)1/2 |
16.742 |