Jump to
S1C2
Energy calculated at B3PW91/3-21G
| | hartrees |
|---|
| Energy at 0K | -356.337652 |
| Energy at 298.15K | -356.343049 |
| Nuclear repulsion energy | 230.766491 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at B3PW91/3-21G
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3690 |
3546 |
76.16 |
|
|
|
| 2 |
A' |
3547 |
3409 |
60.37 |
|
|
|
| 3 |
A' |
3524 |
3387 |
39.36 |
|
|
|
| 4 |
A' |
1801 |
1731 |
149.22 |
|
|
|
| 5 |
A' |
1768 |
1699 |
196.24 |
|
|
|
| 6 |
A' |
1639 |
1575 |
180.52 |
|
|
|
| 7 |
A' |
1425 |
1370 |
11.57 |
|
|
|
| 8 |
A' |
1335 |
1283 |
71.11 |
|
|
|
| 9 |
A' |
1137 |
1093 |
252.21 |
|
|
|
| 10 |
A' |
1116 |
1072 |
18.95 |
|
|
|
| 11 |
A' |
752 |
723 |
6.17 |
|
|
|
| 12 |
A' |
607 |
583 |
64.31 |
|
|
|
| 13 |
A' |
537 |
516 |
2.84 |
|
|
|
| 14 |
A' |
420 |
404 |
10.08 |
|
|
|
| 15 |
A' |
264 |
254 |
14.36 |
|
|
|
| 16 |
A" |
865 |
832 |
2.57 |
|
|
|
| 17 |
A" |
695 |
668 |
501.26 |
|
|
|
| 18 |
A" |
675 |
648 |
1.19 |
|
|
|
| 19 |
A" |
631 |
606 |
11.81 |
|
|
|
| 20 |
A" |
439 |
422 |
41.30 |
|
|
|
| 21 |
A" |
119 |
114 |
7.04 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 13490.7 cm
-1
Scaled (by 0.9612) Zero Point Vibrational Energy (zpe) 12967.2 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at B3PW91/3-21G
Point Group is Cs
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.000 |
0.740 |
0.000 |
| C2 |
-0.055 |
-0.793 |
0.000 |
| O3 |
-1.099 |
-1.446 |
0.000 |
| O4 |
1.045 |
1.387 |
0.000 |
| O5 |
-1.242 |
1.282 |
0.000 |
| N6 |
1.214 |
-1.267 |
0.000 |
| H7 |
1.387 |
-2.264 |
0.000 |
| H8 |
1.979 |
-0.601 |
0.000 |
| H9 |
-1.165 |
2.274 |
0.000 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
O3 |
O4 |
O5 |
N6 |
H7 |
H8 |
H9 |
| C1 | | 1.5337 | 2.4470 | 1.2289 | 1.3548 | 2.3458 | 3.3087 | 2.3908 | 1.9262 |
C2 | 1.5337 | | 1.2315 | 2.4414 | 2.3900 | 1.3550 | 2.0602 | 2.0435 | 3.2614 | O3 | 2.4470 | 1.2315 | | 3.5529 | 2.7321 | 2.3199 | 2.6170 | 3.1922 | 3.7211 | O4 | 1.2289 | 2.4414 | 3.5529 | | 2.2889 | 2.6596 | 3.6668 | 2.1966 | 2.3811 | O5 | 1.3548 | 2.3900 | 2.7321 | 2.2889 | | 3.5396 | 4.4140 | 3.7310 | 0.9951 | N6 | 2.3458 | 1.3550 | 2.3199 | 2.6596 | 3.5396 | | 1.0115 | 1.0147 | 4.2662 | H7 | 3.3087 | 2.0602 | 2.6170 | 3.6668 | 4.4140 | 1.0115 | | 1.7652 | 5.2063 | H8 | 2.3908 | 2.0435 | 3.1922 | 2.1966 | 3.7310 | 1.0147 | 1.7652 | | 4.2605 | H9 | 1.9262 | 3.2614 | 3.7211 | 2.3811 | 0.9951 | 4.2662 | 5.2063 | 4.2605 | |
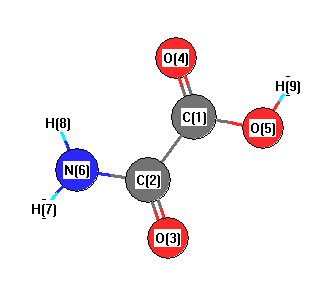 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
O3 |
124.119 |
|
C1 |
C2 |
N6 |
108.440 |
| C1 |
O5 |
H9 |
109.152 |
|
C2 |
C1 |
O4 |
123.827 |
| C2 |
C1 |
O5 |
111.519 |
|
C2 |
N6 |
H7 |
120.353 |
| C2 |
N6 |
H8 |
118.456 |
|
O3 |
C2 |
N6 |
127.441 |
| O4 |
C1 |
O5 |
124.655 |
|
H7 |
N6 |
H8 |
121.191 |
Electronic energy levels
Electronic state
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B3PW91/3-21G
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
0.614 |
|
|
|
| 2 |
C |
0.605 |
|
|
|
| 3 |
O |
-0.485 |
|
|
|
| 4 |
O |
-0.496 |
|
|
|
| 5 |
O |
-0.545 |
|
|
|
| 6 |
N |
-0.787 |
|
|
|
| 7 |
H |
0.348 |
|
|
|
| 8 |
H |
0.357 |
|
|
|
| 9 |
H |
0.389 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
2.265 |
0.947 |
0.000 |
2.455 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-23.499 |
-7.962 |
-0.004 |
| y |
-7.962 |
-38.624 |
0.003 |
| z |
-0.004 |
0.003 |
-32.961 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
12.294 |
-7.962 |
-0.004 |
| y |
-7.962 |
-10.394 |
0.003 |
| z |
-0.004 |
0.003 |
-1.899 |
|
| Polar |
|---|
| 3z2-r2 | -3.799 |
|---|
| x2-y2 | 15.125 |
|---|
| xy | -7.962 |
|---|
| xz | -0.004 |
|---|
| yz | 0.003 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
0.000 |
0.000 |
0.000 |
| y |
0.000 |
0.000 |
0.000 |
| z |
0.000 |
0.000 |
0.000 |
<r2> (average value of r
2) Å
2
| <r2> |
142.274 |
| (<r2>)1/2 |
11.928 |
Jump to
S1C1
Energy calculated at B3PW91/3-21G
| | hartrees |
|---|
| Energy at 0K | -356.346174 |
| Energy at 298.15K | -356.351801 |
| Nuclear repulsion energy | 232.584150 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at B3PW91/3-21G
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3670 |
3528 |
83.44 |
|
|
|
| 2 |
A' |
3532 |
3395 |
71.12 |
|
|
|
| 3 |
A' |
3230 |
3105 |
127.17 |
|
|
|
| 4 |
A' |
1843 |
1771 |
168.24 |
|
|
|
| 5 |
A' |
1773 |
1704 |
156.49 |
|
|
|
| 6 |
A' |
1644 |
1580 |
100.26 |
|
|
|
| 7 |
A' |
1426 |
1371 |
140.22 |
|
|
|
| 8 |
A' |
1352 |
1300 |
449.92 |
|
|
|
| 9 |
A' |
1193 |
1147 |
8.58 |
|
|
|
| 10 |
A' |
1124 |
1080 |
5.48 |
|
|
|
| 11 |
A' |
772 |
742 |
11.24 |
|
|
|
| 12 |
A' |
634 |
609 |
7.34 |
|
|
|
| 13 |
A' |
560 |
538 |
0.92 |
|
|
|
| 14 |
A' |
409 |
393 |
10.37 |
|
|
|
| 15 |
A' |
268 |
258 |
51.62 |
|
|
|
| 16 |
A" |
858 |
825 |
64.25 |
|
|
|
| 17 |
A" |
827 |
795 |
102.39 |
|
|
|
| 18 |
A" |
724 |
696 |
267.99 |
|
|
|
| 19 |
A" |
678 |
651 |
87.25 |
|
|
|
| 20 |
A" |
459 |
441 |
1.06 |
|
|
|
| 21 |
A" |
166 |
159 |
7.78 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 13569.7 cm
-1
Scaled (by 0.9612) Zero Point Vibrational Energy (zpe) 13043.2 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at B3PW91/3-21G
Point Group is Cs
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.031 |
-0.791 |
0.000 |
| C2 |
0.000 |
0.746 |
0.000 |
| O3 |
-1.110 |
1.315 |
0.000 |
| O4 |
1.057 |
-1.456 |
0.000 |
| O5 |
-1.233 |
-1.264 |
0.000 |
| N6 |
1.222 |
1.286 |
0.000 |
| H7 |
1.359 |
2.289 |
0.000 |
| H8 |
2.015 |
0.650 |
0.000 |
| H9 |
-1.819 |
-0.436 |
0.000 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
O3 |
O4 |
O5 |
N6 |
H7 |
H8 |
H9 |
| C1 | | 1.5365 | 2.3950 | 1.2231 | 1.3494 | 2.3939 | 3.3539 | 2.4521 | 1.8836 |
C2 | 1.5365 | | 1.2479 | 2.4421 | 2.3575 | 1.3361 | 2.0564 | 2.0175 | 2.1695 | O3 | 2.3950 | 1.2479 | | 3.5180 | 2.5819 | 2.3325 | 2.6543 | 3.1955 | 1.8895 | O4 | 1.2231 | 2.4421 | 3.5180 | | 2.2982 | 2.7466 | 3.7570 | 2.3132 | 3.0518 | O5 | 1.3494 | 2.3575 | 2.5819 | 2.2982 | | 3.5394 | 4.3978 | 3.7699 | 1.0142 | N6 | 2.3939 | 1.3361 | 2.3325 | 2.7466 | 3.5394 | | 1.0125 | 1.0168 | 3.4950 | H7 | 3.3539 | 2.0564 | 2.6543 | 3.7570 | 4.3978 | 1.0125 | | 1.7659 | 4.1867 | H8 | 2.4521 | 2.0175 | 3.1955 | 2.3132 | 3.7699 | 1.0168 | 1.7659 | | 3.9853 | H9 | 1.8836 | 2.1695 | 1.8895 | 3.0518 | 1.0142 | 3.4950 | 4.1867 | 3.9853 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
O3 |
118.302 |
|
C1 |
C2 |
N6 |
112.707 |
| C1 |
O5 |
H9 |
104.790 |
|
C2 |
C1 |
O4 |
124.088 |
| C2 |
C1 |
O5 |
109.383 |
|
C2 |
N6 |
H7 |
121.622 |
| C2 |
N6 |
H8 |
117.420 |
|
O3 |
C2 |
N6 |
128.991 |
| O4 |
C1 |
O5 |
126.529 |
|
H7 |
N6 |
H8 |
120.959 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B3PW91/3-21G
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
0.657 |
|
|
|
| 2 |
C |
0.587 |
|
|
|
| 3 |
O |
-0.516 |
|
|
|
| 4 |
O |
-0.488 |
|
|
|
| 5 |
O |
-0.570 |
|
|
|
| 6 |
N |
-0.791 |
|
|
|
| 7 |
H |
0.357 |
|
|
|
| 8 |
H |
0.368 |
|
|
|
| 9 |
H |
0.397 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
1.765 |
3.078 |
0.000 |
3.549 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-30.317 |
6.826 |
0.000 |
| y |
6.826 |
-37.022 |
0.000 |
| z |
0.000 |
0.000 |
-32.923 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
4.655 |
6.826 |
0.000 |
| y |
6.826 |
-5.402 |
0.000 |
| z |
0.000 |
0.000 |
0.747 |
|
| Polar |
|---|
| 3z2-r2 | 1.494 |
|---|
| x2-y2 | 6.705 |
|---|
| xy | 6.826 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
6.436 |
-0.005 |
0.000 |
| y |
-0.005 |
4.717 |
0.000 |
| z |
0.000 |
0.000 |
1.624 |
<r2> (average value of r
2) Å
2
| <r2> |
139.597 |
| (<r2>)1/2 |
11.815 |