Jump to
S1C2
Energy calculated at B3PW91/3-21G
| | hartrees |
|---|
| Energy at 0K | -191.974322 |
| Energy at 298.15K | -191.981570 |
| HF Energy | -191.974322 |
| Nuclear repulsion energy | 125.709603 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at B3PW91/3-21G
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A1 |
3116 |
2995 |
17.75 |
|
|
|
| 2 |
A1 |
3077 |
2958 |
5.18 |
|
|
|
| 3 |
A1 |
1591 |
1529 |
0.21 |
|
|
|
| 4 |
A1 |
1525 |
1466 |
4.69 |
|
|
|
| 5 |
A1 |
1365 |
1312 |
0.69 |
|
|
|
| 6 |
A1 |
1016 |
977 |
0.07 |
|
|
|
| 7 |
A1 |
875 |
841 |
11.51 |
|
|
|
| 8 |
A1 |
804 |
773 |
5.66 |
|
|
|
| 9 |
A2 |
3123 |
3002 |
0.00 |
|
|
|
| 10 |
A2 |
1272 |
1222 |
0.00 |
|
|
|
| 11 |
A2 |
1135 |
1091 |
0.00 |
|
|
|
| 12 |
A2 |
877 |
843 |
0.00 |
|
|
|
| 13 |
B1 |
3179 |
3055 |
30.91 |
|
|
|
| 14 |
B1 |
3122 |
3001 |
60.89 |
|
|
|
| 15 |
B1 |
1210 |
1163 |
2.22 |
|
|
|
| 16 |
B1 |
1096 |
1054 |
0.15 |
|
|
|
| 17 |
B1 |
767 |
737 |
1.74 |
|
|
|
| 18 |
B1 |
94 |
90 |
3.05 |
|
|
|
| 19 |
B2 |
3067 |
2948 |
114.85 |
|
|
|
| 20 |
B2 |
1560 |
1499 |
1.06 |
|
|
|
| 21 |
B2 |
1307 |
1257 |
22.00 |
|
|
|
| 22 |
B2 |
1280 |
1230 |
2.24 |
|
|
|
| 23 |
B2 |
990 |
952 |
48.02 |
|
|
|
| 24 |
B2 |
930 |
894 |
19.11 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 19188.0 cm
-1
Scaled (by 0.9612) Zero Point Vibrational Energy (zpe) 18443.5 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at B3PW91/3-21G
Point Group is C2v
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| O1 |
0.000 |
0.000 |
1.109 |
| C2 |
0.000 |
0.000 |
-1.081 |
| C3 |
0.000 |
1.066 |
0.052 |
| C4 |
0.000 |
-1.066 |
0.052 |
| H5 |
0.896 |
0.000 |
-1.704 |
| H6 |
-0.896 |
0.000 |
-1.704 |
| H7 |
0.899 |
1.688 |
0.101 |
| H8 |
-0.899 |
1.688 |
0.101 |
| H9 |
-0.899 |
-1.688 |
0.101 |
| H10 |
0.899 |
-1.688 |
0.101 |
Atom - Atom Distances (Å)
| |
O1 |
C2 |
C3 |
C4 |
H5 |
H6 |
H7 |
H8 |
H9 |
H10 |
| O1 | | 2.1897 | 1.5015 | 1.5015 | 2.9525 | 2.9525 | 2.1616 | 2.1616 | 2.1616 | 2.1616 |
C2 | 2.1897 | | 1.5555 | 1.5555 | 1.0919 | 1.0919 | 2.2482 | 2.2482 | 2.2482 | 2.2482 | C3 | 1.5015 | 1.5555 | | 2.1326 | 2.2414 | 2.2414 | 1.0939 | 1.0939 | 2.8977 | 2.8977 | C4 | 1.5015 | 1.5555 | 2.1326 | | 2.2414 | 2.2414 | 2.8977 | 2.8977 | 1.0939 | 1.0939 | H5 | 2.9525 | 1.0919 | 2.2414 | 2.2414 | | 1.7927 | 2.4717 | 3.0548 | 3.0548 | 2.4717 | H6 | 2.9525 | 1.0919 | 2.2414 | 2.2414 | 1.7927 | | 3.0548 | 2.4717 | 2.4717 | 3.0548 | H7 | 2.1616 | 2.2482 | 1.0939 | 2.8977 | 2.4717 | 3.0548 | | 1.7974 | 3.8247 | 3.3761 | H8 | 2.1616 | 2.2482 | 1.0939 | 2.8977 | 3.0548 | 2.4717 | 1.7974 | | 3.3761 | 3.8247 | H9 | 2.1616 | 2.2482 | 2.8977 | 1.0939 | 3.0548 | 2.4717 | 3.8247 | 3.3761 | | 1.7974 | H10 | 2.1616 | 2.2482 | 2.8977 | 1.0939 | 2.4717 | 3.0548 | 3.3761 | 3.8247 | 1.7974 | |
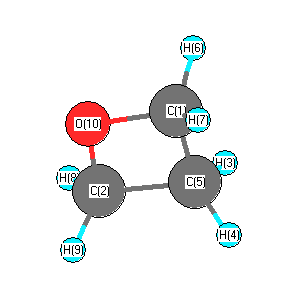 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| O1 |
C3 |
C2 |
91.479 |
|
O1 |
C3 |
H7 |
111.823 |
| O1 |
C3 |
H8 |
111.823 |
|
O1 |
C4 |
C2 |
91.479 |
| O1 |
C4 |
H9 |
111.823 |
|
O1 |
C4 |
H10 |
111.823 |
| C2 |
C3 |
H7 |
114.999 |
|
C2 |
C3 |
H8 |
114.999 |
| C2 |
C4 |
H9 |
114.999 |
|
C2 |
C4 |
H10 |
114.999 |
| C3 |
O1 |
C4 |
90.490 |
|
C3 |
C2 |
C4 |
86.551 |
| C3 |
C2 |
H5 |
114.567 |
|
C3 |
C2 |
H6 |
114.567 |
| C4 |
C2 |
H5 |
114.567 |
|
C4 |
C2 |
H6 |
114.567 |
| H5 |
C2 |
H6 |
110.355 |
|
H7 |
C3 |
H8 |
110.473 |
| H9 |
C4 |
H10 |
110.473 |
|
Electronic energy levels
Electronic state
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B3PW91/3-21G
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
O |
-0.464 |
|
|
|
| 2 |
C |
-0.534 |
|
|
|
| 3 |
C |
-0.162 |
|
|
|
| 4 |
C |
-0.162 |
|
|
|
| 5 |
H |
0.231 |
|
|
|
| 6 |
H |
0.231 |
|
|
|
| 7 |
H |
0.215 |
|
|
|
| 8 |
H |
0.215 |
|
|
|
| 9 |
H |
0.215 |
|
|
|
| 10 |
H |
0.215 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
-2.300 |
2.300 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-23.991 |
0.000 |
0.000 |
| y |
0.000 |
-22.322 |
0.000 |
| z |
0.000 |
0.000 |
-27.929 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
1.135 |
0.000 |
0.000 |
| y |
0.000 |
3.638 |
0.000 |
| z |
0.000 |
0.000 |
-4.773 |
|
| Polar |
|---|
| 3z2-r2 | -9.545 |
|---|
| x2-y2 | -1.669 |
|---|
| xy | 0.000 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
4.307 |
0.000 |
0.000 |
| y |
0.000 |
5.454 |
0.000 |
| z |
0.000 |
0.000 |
4.135 |
<r2> (average value of r
2) Å
2
| <r2> |
68.064 |
| (<r2>)1/2 |
8.250 |
Jump to
S1C1
Energy calculated at B3PW91/3-21G
| | hartrees |
|---|
| Energy at 0K | -191.974322 |
| Energy at 298.15K | -191.981571 |
| HF Energy | -191.974322 |
| Nuclear repulsion energy | 125.707666 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at B3PW91/3-21G
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3178 |
3055 |
31.16 |
|
|
|
| 2 |
A' |
3122 |
3001 |
60.64 |
|
|
|
| 3 |
A' |
3115 |
2994 |
17.74 |
|
|
|
| 4 |
A' |
3077 |
2958 |
5.21 |
|
|
|
| 5 |
A' |
1591 |
1529 |
0.21 |
|
|
|
| 6 |
A' |
1525 |
1466 |
4.68 |
|
|
|
| 7 |
A' |
1365 |
1312 |
0.69 |
|
|
|
| 8 |
A' |
1210 |
1163 |
2.23 |
|
|
|
| 9 |
A' |
1096 |
1053 |
0.14 |
|
|
|
| 10 |
A' |
1016 |
977 |
0.06 |
|
|
|
| 11 |
A' |
875 |
841 |
11.49 |
|
|
|
| 12 |
A' |
804 |
773 |
5.67 |
|
|
|
| 13 |
A' |
767 |
737 |
1.75 |
|
|
|
| 14 |
A' |
94 |
90 |
3.05 |
|
|
|
| 15 |
A" |
3123 |
3001 |
0.00 |
|
|
|
| 16 |
A" |
3067 |
2948 |
114.83 |
|
|
|
| 17 |
A" |
1560 |
1499 |
1.05 |
|
|
|
| 18 |
A" |
1308 |
1257 |
22.06 |
|
|
|
| 19 |
A" |
1280 |
1230 |
2.16 |
|
|
|
| 20 |
A" |
1272 |
1222 |
0.00 |
|
|
|
| 21 |
A" |
1135 |
1091 |
0.00 |
|
|
|
| 22 |
A" |
990 |
951 |
47.63 |
|
|
|
| 23 |
A" |
930 |
894 |
19.46 |
|
|
|
| 24 |
A" |
877 |
843 |
0.00 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 19187.9 cm
-1
Scaled (by 0.9612) Zero Point Vibrational Energy (zpe) 18443.4 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at B3PW91/3-21G
Point Group is Cs
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| O1 |
0.002 |
-1.109 |
0.000 |
| C2 |
-0.001 |
1.081 |
0.000 |
| C3 |
-0.001 |
-0.051 |
1.066 |
| C4 |
-0.001 |
-0.051 |
-1.066 |
| H5 |
0.896 |
1.704 |
0.000 |
| H6 |
-0.896 |
1.705 |
0.000 |
| H7 |
0.897 |
-0.100 |
1.689 |
| H8 |
-0.900 |
-0.102 |
1.687 |
| H9 |
0.897 |
-0.100 |
-1.689 |
| H10 |
-0.900 |
-0.102 |
-1.687 |
Atom - Atom Distances (Å)
| |
O1 |
C2 |
C3 |
C4 |
H5 |
H6 |
H7 |
H8 |
H9 |
H10 |
| O1 | | 2.1898 | 1.5017 | 1.5017 | 2.9518 | 2.9540 | 2.1617 | 2.1617 | 2.1617 | 2.1617 |
C2 | 2.1898 | | 1.5553 | 1.5553 | 1.0919 | 1.0920 | 2.2484 | 2.2479 | 2.2484 | 2.2479 | C3 | 1.5017 | 1.5553 | | 2.1324 | 2.2412 | 2.2416 | 1.0940 | 1.0940 | 2.8986 | 2.8966 | C4 | 1.5017 | 1.5553 | 2.1324 | | 2.2412 | 2.2416 | 2.8986 | 2.8966 | 1.0940 | 1.0940 | H5 | 2.9518 | 1.0919 | 2.2412 | 2.2412 | | 1.7924 | 2.4719 | 3.0550 | 2.4719 | 3.0550 | H6 | 2.9540 | 1.0920 | 2.2416 | 2.2416 | 1.7924 | | 3.0546 | 2.4719 | 3.0546 | 2.4719 | H7 | 2.1617 | 2.2484 | 1.0940 | 2.8986 | 2.4719 | 3.0546 | | 1.7973 | 3.3788 | 3.8247 | H8 | 2.1617 | 2.2479 | 1.0940 | 2.8966 | 3.0550 | 2.4719 | 1.7973 | | 3.8247 | 3.3735 | H9 | 2.1617 | 2.2484 | 2.8986 | 1.0940 | 2.4719 | 3.0546 | 3.3788 | 3.8247 | | 1.7973 | H10 | 2.1617 | 2.2479 | 2.8966 | 1.0940 | 3.0550 | 2.4719 | 3.8247 | 3.3735 | 1.7973 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| O1 |
C3 |
C2 |
91.486 |
|
O1 |
C3 |
H7 |
111.817 |
| O1 |
C3 |
H8 |
111.816 |
|
O1 |
C4 |
C2 |
91.486 |
| O1 |
C4 |
H9 |
111.817 |
|
O1 |
C4 |
H10 |
111.816 |
| C2 |
C3 |
H7 |
115.028 |
|
C2 |
C3 |
H8 |
114.989 |
| C2 |
C4 |
H9 |
115.028 |
|
C2 |
C4 |
H10 |
114.989 |
| C3 |
O1 |
C4 |
90.469 |
|
C3 |
C2 |
C4 |
86.559 |
| C3 |
C2 |
H5 |
114.558 |
|
C3 |
C2 |
H6 |
114.596 |
| C4 |
C2 |
H5 |
114.558 |
|
C4 |
C2 |
H6 |
114.596 |
| H5 |
C2 |
H6 |
110.318 |
|
H7 |
C3 |
H8 |
110.463 |
| H9 |
C4 |
H10 |
110.463 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B3PW91/3-21G
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
O |
-0.464 |
|
|
|
| 2 |
C |
-0.534 |
|
|
|
| 3 |
C |
-0.162 |
|
|
|
| 4 |
C |
-0.162 |
|
|
|
| 5 |
H |
0.231 |
|
|
|
| 6 |
H |
0.231 |
|
|
|
| 7 |
H |
0.215 |
|
|
|
| 8 |
H |
0.215 |
|
|
|
| 9 |
H |
0.215 |
|
|
|
| 10 |
H |
0.215 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
-0.004 |
2.301 |
0.000 |
2.301 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-23.992 |
0.006 |
0.000 |
| y |
0.006 |
-27.928 |
0.000 |
| z |
0.000 |
0.000 |
-22.322 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
1.132 |
0.006 |
0.000 |
| y |
0.006 |
-4.771 |
0.000 |
| z |
0.000 |
0.000 |
3.639 |
|
| Polar |
|---|
| 3z2-r2 | 7.277 |
|---|
| x2-y2 | 3.936 |
|---|
| xy | 0.006 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
4.307 |
-0.000 |
0.000 |
| y |
-0.000 |
4.135 |
0.000 |
| z |
0.000 |
0.000 |
5.454 |
<r2> (average value of r
2) Å
2
| <r2> |
68.065 |
| (<r2>)1/2 |
8.250 |