Jump to
S1C2
Vibrational Frequencies calculated at B3PW91/TZVP
Geometric Data calculated at B3PW91/TZVP
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Jump to
S1C1
Energy calculated at B3PW91/TZVP
| | hartrees |
|---|
| Energy at 0K | -261.038593 |
| Energy at 298.15K | -261.043616 |
| HF Energy | -261.038593 |
| Nuclear repulsion energy | 127.713796 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at B3PW91/TZVP
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3549 |
3422 |
40.71 |
|
|
|
| 2 |
A' |
1610 |
1553 |
73.82 |
|
|
|
| 3 |
A' |
1397 |
1347 |
266.61 |
|
|
|
| 4 |
A' |
1025 |
988 |
30.01 |
|
|
|
| 5 |
A' |
809 |
780 |
74.59 |
|
|
|
| 6 |
A' |
728 |
702 |
66.44 |
|
|
|
| 7 |
A' |
612 |
590 |
199.41 |
|
|
|
| 8 |
A" |
3688 |
3556 |
57.77 |
|
|
|
| 9 |
A" |
1688 |
1628 |
377.79 |
|
|
|
| 10 |
A" |
1235 |
1191 |
50.48 |
|
|
|
| 11 |
A" |
576 |
555 |
1.63 |
|
|
|
| 12 |
A" |
430 |
415 |
22.94 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 8673.7 cm
-1
Scaled (by 0.9643) Zero Point Vibrational Energy (zpe) 8364.1 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at B3PW91/TZVP
Point Group is Cs
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| N1 |
0.075 |
-1.240 |
0.000 |
| N2 |
0.002 |
0.143 |
0.000 |
| O3 |
0.002 |
0.682 |
1.088 |
| O4 |
0.002 |
0.682 |
-1.088 |
| H5 |
-0.292 |
-1.619 |
-0.862 |
| H6 |
-0.292 |
-1.619 |
0.862 |
Atom - Atom Distances (Å)
| |
N1 |
N2 |
O3 |
O4 |
H5 |
H6 |
| N1 | | 1.3854 | 2.2104 | 2.2104 | 1.0105 | 1.0105 |
N2 | 1.3854 | | 1.2141 | 1.2141 | 1.9836 | 1.9836 | O3 | 2.2104 | 1.2141 | | 2.1754 | 3.0305 | 2.3315 | O4 | 2.2104 | 1.2141 | 2.1754 | | 2.3315 | 3.0305 | H5 | 1.0105 | 1.9836 | 3.0305 | 2.3315 | | 1.7231 | H6 | 1.0105 | 1.9836 | 2.3315 | 3.0305 | 1.7231 | |
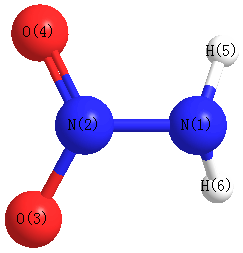 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| N1 |
N2 |
O3 |
116.337 |
|
N1 |
N2 |
O4 |
116.337 |
| N2 |
N1 |
H5 |
110.805 |
|
N2 |
N1 |
H6 |
110.805 |
| O3 |
N2 |
O4 |
127.247 |
|
H5 |
N1 |
H6 |
116.995 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B3PW91/TZVP
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
N |
-0.310 |
|
|
|
| 2 |
N |
0.212 |
|
|
|
| 3 |
O |
-0.202 |
|
|
|
| 4 |
O |
-0.202 |
|
|
|
| 5 |
H |
0.252 |
|
|
|
| 6 |
H |
0.252 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
-1.167 |
-3.797 |
0.000 |
3.973 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-22.382 |
2.215 |
0.000 |
| y |
2.215 |
-20.373 |
0.000 |
| z |
0.000 |
0.000 |
-24.376 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-0.008 |
2.215 |
0.000 |
| y |
2.215 |
3.007 |
0.000 |
| z |
0.000 |
0.000 |
-2.999 |
|
| Polar |
|---|
| 3z2-r2 | -5.998 |
|---|
| x2-y2 | -2.010 |
|---|
| xy | 2.215 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
2.147 |
0.015 |
0.000 |
| y |
0.015 |
4.394 |
0.000 |
| z |
0.000 |
0.000 |
4.850 |
<r2> (average value of r
2) Å
2
| <r2> |
58.210 |
| (<r2>)1/2 |
7.630 |