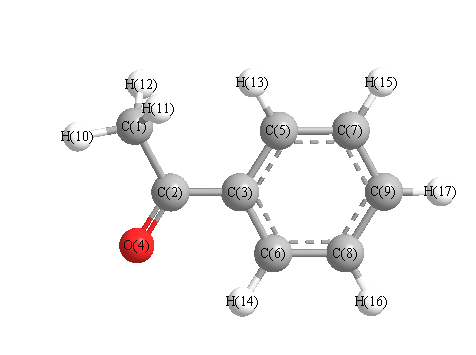
Vibrational Frequencies calculated at B3PW91/TZVP
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3211 |
3097 |
7.14 |
|
|
|
| 2 |
A' |
3206 |
3092 |
8.74 |
|
|
|
| 3 |
A' |
3198 |
3084 |
12.90 |
|
|
|
| 4 |
A' |
3189 |
3075 |
5.58 |
|
|
|
| 5 |
A' |
3179 |
3065 |
0.10 |
|
|
|
| 6 |
A' |
3164 |
3051 |
10.14 |
|
|
|
| 7 |
A' |
3048 |
2939 |
2.09 |
|
|
|
| 8 |
A' |
1771 |
1708 |
195.36 |
|
|
|
| 9 |
A' |
1652 |
1593 |
21.71 |
|
|
|
| 10 |
A' |
1634 |
1575 |
8.65 |
|
|
|
| 11 |
A' |
1526 |
1471 |
0.62 |
|
|
|
| 12 |
A' |
1484 |
1431 |
18.42 |
|
|
|
| 13 |
A' |
1465 |
1413 |
11.30 |
|
|
|
| 14 |
A' |
1383 |
1334 |
63.39 |
|
|
|
| 15 |
A' |
1366 |
1317 |
3.43 |
|
|
|
| 16 |
A' |
1341 |
1293 |
3.03 |
|
|
|
| 17 |
A' |
1281 |
1235 |
182.06 |
|
|
|
| 18 |
A' |
1202 |
1160 |
16.27 |
|
|
|
| 19 |
A' |
1187 |
1145 |
0.67 |
|
|
|
| 20 |
A' |
1110 |
1071 |
7.35 |
|
|
|
| 21 |
A' |
1096 |
1057 |
2.23 |
|
|
|
| 22 |
A' |
1051 |
1014 |
7.13 |
|
|
|
| 23 |
A' |
1021 |
984 |
0.95 |
|
|
|
| 24 |
A' |
958 |
924 |
28.36 |
|
|
|
| 25 |
A' |
745 |
718 |
0.84 |
|
|
|
| 26 |
A' |
631 |
608 |
0.98 |
|
|
|
| 27 |
A' |
599 |
578 |
24.63 |
|
|
|
| 28 |
A' |
466 |
449 |
0.75 |
|
|
|
| 29 |
A' |
365 |
352 |
0.65 |
|
|
|
| 30 |
A' |
215 |
207 |
5.02 |
|
|
|
| 31 |
A" |
3115 |
3004 |
5.91 |
|
|
|
| 32 |
A" |
1474 |
1421 |
13.60 |
|
|
|
| 33 |
A" |
1040 |
1003 |
0.67 |
|
|
|
| 34 |
A" |
1000 |
965 |
0.41 |
|
|
|
| 35 |
A" |
984 |
949 |
0.21 |
|
|
|
| 36 |
A" |
939 |
905 |
2.02 |
|
|
|
| 37 |
A" |
864 |
834 |
0.18 |
|
|
|
| 38 |
A" |
763 |
736 |
55.94 |
|
|
|
| 39 |
A" |
680 |
656 |
25.04 |
|
|
|
| 40 |
A" |
594 |
573 |
9.25 |
|
|
|
| 41 |
A" |
420 |
405 |
0.26 |
|
|
|
| 42 |
A" |
407 |
393 |
0.00 |
|
|
|
| 43 |
A" |
152 |
147 |
0.01 |
|
|
|
| 44 |
A" |
141 |
136 |
0.12 |
|
|
|
| 45 |
A" |
51 |
49 |
3.81 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 30183.4 cm
-1
Scaled (by 0.9643) Zero Point Vibrational Energy (zpe) 29105.9 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B3PW91/TZVP
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.440 |
|
|
|
| 2 |
C |
0.144 |
|
|
|
| 3 |
C |
-0.026 |
|
|
|
| 4 |
O |
-0.279 |
|
|
|
| 5 |
C |
-0.087 |
|
|
|
| 6 |
C |
-0.121 |
|
|
|
| 7 |
C |
-0.108 |
|
|
|
| 8 |
C |
-0.090 |
|
|
|
| 9 |
C |
-0.101 |
|
|
|
| 10 |
H |
0.166 |
|
|
|
| 11 |
H |
0.146 |
|
|
|
| 12 |
H |
0.146 |
|
|
|
| 13 |
H |
0.145 |
|
|
|
| 14 |
H |
0.148 |
|
|
|
| 15 |
H |
0.116 |
|
|
|
| 16 |
H |
0.121 |
|
|
|
| 17 |
H |
0.119 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
1.917 |
-2.512 |
0.000 |
3.160 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-47.127 |
5.709 |
0.000 |
| y |
5.709 |
-54.189 |
0.000 |
| z |
0.000 |
0.000 |
-55.272 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
7.603 |
5.709 |
0.000 |
| y |
5.709 |
-2.989 |
0.000 |
| z |
0.000 |
0.000 |
-4.614 |
|
| Polar |
|---|
| 3z2-r2 | -9.228 |
|---|
| x2-y2 | 7.062 |
|---|
| xy | 5.709 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
14.825 |
0.693 |
0.000 |
| y |
0.693 |
17.944 |
0.000 |
| z |
0.000 |
0.000 |
7.726 |
<r2> (average value of r
2) Å
2
| <r2> |
344.074 |
| (<r2>)1/2 |
18.549 |