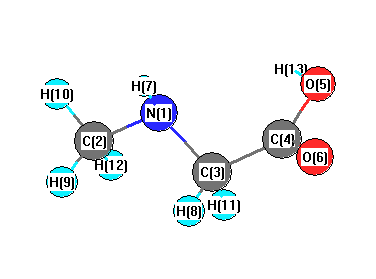
Vibrational Frequencies calculated at B3PW91/6-31+G**
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3565 |
3422 |
6.78 |
|
|
|
| 2 |
A |
3415 |
3278 |
388.01 |
|
|
|
| 3 |
A |
3146 |
3021 |
18.57 |
|
|
|
| 4 |
A |
3115 |
2991 |
3.90 |
|
|
|
| 5 |
A |
3104 |
2980 |
26.54 |
|
|
|
| 6 |
A |
3062 |
2939 |
16.22 |
|
|
|
| 7 |
A |
3010 |
2890 |
77.87 |
|
|
|
| 8 |
A |
1870 |
1795 |
420.35 |
|
|
|
| 9 |
A |
1519 |
1459 |
16.63 |
|
|
|
| 10 |
A |
1498 |
1438 |
18.72 |
|
|
|
| 11 |
A |
1489 |
1430 |
17.37 |
|
|
|
| 12 |
A |
1473 |
1414 |
12.52 |
|
|
|
| 13 |
A |
1454 |
1396 |
4.91 |
|
|
|
| 14 |
A |
1427 |
1370 |
391.05 |
|
|
|
| 15 |
A |
1344 |
1290 |
9.14 |
|
|
|
| 16 |
A |
1291 |
1239 |
2.86 |
|
|
|
| 17 |
A |
1232 |
1183 |
17.30 |
|
|
|
| 18 |
A |
1184 |
1137 |
33.30 |
|
|
|
| 19 |
A |
1158 |
1112 |
25.88 |
|
|
|
| 20 |
A |
1139 |
1093 |
11.67 |
|
|
|
| 21 |
A |
1011 |
970 |
24.74 |
|
|
|
| 22 |
A |
972 |
933 |
30.60 |
|
|
|
| 23 |
A |
926 |
889 |
50.52 |
|
|
|
| 24 |
A |
883 |
848 |
19.93 |
|
|
|
| 25 |
A |
785 |
754 |
67.49 |
|
|
|
| 26 |
A |
636 |
611 |
3.76 |
|
|
|
| 27 |
A |
571 |
548 |
8.55 |
|
|
|
| 28 |
A |
484 |
464 |
8.99 |
|
|
|
| 29 |
A |
374 |
359 |
4.94 |
|
|
|
| 30 |
A |
296 |
284 |
9.65 |
|
|
|
| 31 |
A |
205 |
197 |
1.99 |
|
|
|
| 32 |
A |
135 |
129 |
3.08 |
|
|
|
| 33 |
A |
48 |
46 |
5.46 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 23909.0 cm
-1
Scaled (by 0.9601) Zero Point Vibrational Energy (zpe) 22955.0 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B3PW91/6-31+G**
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
N |
-0.506 |
|
|
|
| 2 |
C |
-0.380 |
|
|
|
| 3 |
C |
-0.230 |
|
|
|
| 4 |
C |
0.399 |
|
|
|
| 5 |
O |
-0.421 |
|
|
|
| 6 |
O |
-0.456 |
|
|
|
| 7 |
H |
0.323 |
|
|
|
| 8 |
H |
0.194 |
|
|
|
| 9 |
H |
0.156 |
|
|
|
| 10 |
H |
0.168 |
|
|
|
| 11 |
H |
0.194 |
|
|
|
| 12 |
H |
0.169 |
|
|
|
| 13 |
H |
0.390 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
-5.845 |
-0.401 |
0.808 |
5.914 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-44.023 |
0.705 |
-0.149 |
| y |
0.705 |
-38.007 |
0.376 |
| z |
-0.149 |
0.376 |
-33.452 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-8.293 |
0.705 |
-0.149 |
| y |
0.705 |
0.730 |
0.376 |
| z |
-0.149 |
0.376 |
7.563 |
|
| Polar |
|---|
| 3z2-r2 | 15.126 |
|---|
| x2-y2 | -6.015 |
|---|
| xy | 0.705 |
|---|
| xz | -0.149 |
|---|
| yz | 0.376 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
9.373 |
-0.405 |
0.002 |
| y |
-0.405 |
7.601 |
0.004 |
| z |
0.002 |
0.004 |
5.880 |
<r2> (average value of r
2) Å
2
| <r2> |
0.000 |
| (<r2>)1/2 |
0.000 |