Jump to
S1C2
Energy calculated at B3PW91/6-31+G**
| | hartrees |
|---|
| Energy at 0K | -151.498439 |
| Energy at 298.15K | -151.500747 |
| HF Energy | -151.498439 |
| Nuclear repulsion energy | 36.959168 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at B3PW91/6-31+G**
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3805 |
3653 |
13.17 |
|
|
|
| 2 |
A |
1459 |
1401 |
0.08 |
|
|
|
| 3 |
A |
976 |
937 |
1.13 |
|
|
|
| 4 |
A |
387 |
371 |
222.10 |
|
|
|
| 5 |
B |
3804 |
3653 |
65.51 |
|
|
|
| 6 |
B |
1320 |
1268 |
99.13 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 5875.3 cm
-1
Scaled (by 0.9601) Zero Point Vibrational Energy (zpe) 5640.8 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at B3PW91/6-31+G**
Point Group is C2
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| O1 |
0.000 |
0.721 |
-0.054 |
| O2 |
0.000 |
-0.721 |
-0.054 |
| H3 |
0.816 |
0.899 |
0.435 |
| H4 |
-0.816 |
-0.899 |
0.435 |
Atom - Atom Distances (Å)
| |
O1 |
O2 |
H3 |
H4 |
| O1 | | 1.4415 | 0.9687 | 1.8787 |
O2 | 1.4415 | | 1.8787 | 0.9687 | H3 | 0.9687 | 1.8787 | | 2.4286 | H4 | 1.8787 | 0.9687 | 2.4286 | |
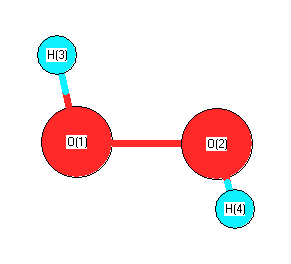 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| O1 |
O2 |
H4 |
100.589 |
|
O2 |
O1 |
H3 |
100.589 |
Electronic energy levels
Electronic state
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B3PW91/6-31+G**
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
O |
-0.366 |
|
|
|
| 2 |
O |
-0.366 |
|
|
|
| 3 |
H |
0.366 |
|
|
|
| 4 |
H |
0.366 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
1.829 |
1.829 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-9.533 |
3.243 |
0.000 |
| y |
3.243 |
-11.577 |
0.000 |
| z |
0.000 |
0.000 |
-12.108 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
2.310 |
3.243 |
0.000 |
| y |
3.243 |
-0.757 |
0.000 |
| z |
0.000 |
0.000 |
-1.554 |
|
| Polar |
|---|
| 3z2-r2 | -3.107 |
|---|
| x2-y2 | 2.045 |
|---|
| xy | 3.243 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
1.633 |
0.223 |
0.000 |
| y |
0.223 |
2.361 |
0.000 |
| z |
0.000 |
0.000 |
1.482 |
<r2> (average value of r
2) Å
2
| <r2> |
18.603 |
| (<r2>)1/2 |
4.313 |
Jump to
S1C1
Energy calculated at B3PW91/6-31+G**
| | hartrees |
|---|
| Energy at 0K | -151.496989 |
| Energy at 298.15K | |
| HF Energy | -151.496989 |
| Nuclear repulsion energy | 36.815126 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at B3PW91/6-31+G**
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
Ag |
3835 |
3682 |
0.00 |
|
|
|
| 2 |
Ag |
1541 |
1480 |
0.00 |
|
|
|
| 3 |
Ag |
974 |
936 |
0.00 |
|
|
|
| 4 |
Au |
299i |
287i |
318.09 |
|
|
|
| 5 |
Bu |
3841 |
3688 |
113.47 |
|
|
|
| 6 |
Bu |
1248 |
1198 |
134.83 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 5570.2 cm
-1
Scaled (by 0.9601) Zero Point Vibrational Energy (zpe) 5347.9 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at B3PW91/6-31+G**
Point Group is C2h
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| O1 |
0.000 |
0.726 |
0.000 |
| O2 |
0.000 |
-0.726 |
0.000 |
| H3 |
0.955 |
0.879 |
0.000 |
| H4 |
-0.955 |
-0.879 |
0.000 |
Atom - Atom Distances (Å)
| |
O1 |
O2 |
H3 |
H4 |
| O1 | | 1.4519 | 0.9675 | 1.8674 |
O2 | 1.4519 | | 1.8674 | 0.9675 | H3 | 0.9675 | 1.8674 | | 2.5958 | H4 | 1.8674 | 0.9675 | 2.5958 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| O1 |
O2 |
H4 |
99.073 |
|
O2 |
O1 |
H3 |
99.073 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B3PW91/6-31+G**
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
O |
-0.372 |
|
|
|
| 2 |
O |
-0.372 |
|
|
|
| 3 |
H |
0.372 |
|
|
|
| 4 |
H |
0.372 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
0.000 |
0.000 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-8.141 |
3.750 |
0.000 |
| y |
3.750 |
-11.704 |
0.000 |
| z |
0.000 |
0.000 |
-13.225 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
4.323 |
3.750 |
0.000 |
| y |
3.750 |
-1.021 |
0.000 |
| z |
0.000 |
0.000 |
-3.302 |
|
| Polar |
|---|
| 3z2-r2 | -6.604 |
|---|
| x2-y2 | 3.563 |
|---|
| xy | 3.750 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
1.711 |
0.240 |
0.000 |
| y |
0.240 |
2.383 |
0.000 |
| z |
0.000 |
0.000 |
1.388 |
<r2> (average value of r
2) Å
2
| <r2> |
18.686 |
| (<r2>)1/2 |
4.323 |