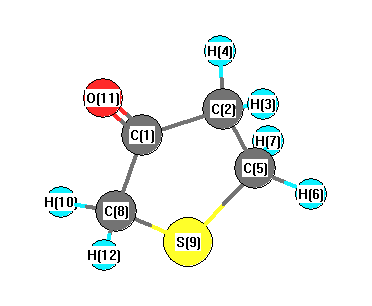
Vibrational Frequencies calculated at B3PW91/cc-pVTZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3132 |
3012 |
4.94 |
|
|
|
| 2 |
A |
3125 |
3005 |
0.59 |
|
|
|
| 3 |
A |
3110 |
2991 |
8.05 |
|
|
|
| 4 |
A |
3061 |
2944 |
25.09 |
|
|
|
| 5 |
A |
3050 |
2933 |
14.12 |
|
|
|
| 6 |
A |
3045 |
2928 |
3.85 |
|
|
|
| 7 |
A |
1827 |
1757 |
232.77 |
|
|
|
| 8 |
A |
1486 |
1429 |
2.49 |
|
|
|
| 9 |
A |
1436 |
1380 |
2.10 |
|
|
|
| 10 |
A |
1433 |
1378 |
17.40 |
|
|
|
| 11 |
A |
1301 |
1251 |
12.35 |
|
|
|
| 12 |
A |
1288 |
1239 |
9.86 |
|
|
|
| 13 |
A |
1223 |
1176 |
5.03 |
|
|
|
| 14 |
A |
1215 |
1168 |
23.98 |
|
|
|
| 15 |
A |
1162 |
1118 |
4.45 |
|
|
|
| 16 |
A |
1142 |
1098 |
50.51 |
|
|
|
| 17 |
A |
1089 |
1047 |
2.28 |
|
|
|
| 18 |
A |
1005 |
966 |
8.78 |
|
|
|
| 19 |
A |
972 |
934 |
0.48 |
|
|
|
| 20 |
A |
869 |
835 |
6.48 |
|
|
|
| 21 |
A |
828 |
796 |
2.24 |
|
|
|
| 22 |
A |
792 |
762 |
0.44 |
|
|
|
| 23 |
A |
729 |
701 |
6.20 |
|
|
|
| 24 |
A |
684 |
658 |
0.71 |
|
|
|
| 25 |
A |
560 |
539 |
4.47 |
|
|
|
| 26 |
A |
490 |
471 |
4.78 |
|
|
|
| 27 |
A |
446 |
429 |
5.42 |
|
|
|
| 28 |
A |
429 |
412 |
3.24 |
|
|
|
| 29 |
A |
186 |
179 |
1.95 |
|
|
|
| 30 |
A |
68 |
66 |
11.16 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 20591.2 cm
-1
Scaled (by 0.9616) Zero Point Vibrational Energy (zpe) 19800.5 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B3PW91/cc-pVTZ
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
0.184 |
|
|
|
| 2 |
C |
-0.176 |
|
|
|
| 3 |
H |
0.111 |
|
|
|
| 4 |
H |
0.119 |
|
|
|
| 5 |
C |
-0.172 |
|
|
|
| 6 |
H |
0.109 |
|
|
|
| 7 |
H |
0.110 |
|
|
|
| 8 |
C |
-0.153 |
|
|
|
| 9 |
S |
-0.111 |
|
|
|
| 10 |
H |
0.120 |
|
|
|
| 11 |
O |
-0.257 |
|
|
|
| 12 |
H |
0.116 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
1.211 |
1.300 |
0.383 |
1.818 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-53.512 |
1.042 |
0.291 |
| y |
1.042 |
-38.332 |
-0.051 |
| z |
0.291 |
-0.051 |
-41.819 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-13.436 |
1.042 |
0.291 |
| y |
1.042 |
9.334 |
-0.051 |
| z |
0.291 |
-0.051 |
4.102 |
|
| Polar |
|---|
| 3z2-r2 | 8.205 |
|---|
| x2-y2 | -15.180 |
|---|
| xy | 1.042 |
|---|
| xz | 0.291 |
|---|
| yz | -0.051 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
0.000 |
0.000 |
0.000 |
| y |
0.000 |
0.000 |
0.000 |
| z |
0.000 |
0.000 |
0.000 |
<r2> (average value of r
2) Å
2
| <r2> |
184.511 |
| (<r2>)1/2 |
13.583 |