Jump to
S1C2
Energy calculated at B3PW91/cc-pVTZ
| | hartrees |
|---|
| Energy at 0K | -275.519493 |
| Energy at 298.15K | |
| HF Energy | -275.519493 |
| Nuclear repulsion energy | 117.655893 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at B3PW91/cc-pVTZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A1 |
2119 |
2037 |
40.82 |
|
|
|
| 2 |
A1 |
745 |
717 |
9.58 |
|
|
|
| 3 |
A1 |
529 |
508 |
88.12 |
|
|
|
| 4 |
A1 |
89 |
86 |
11.89 |
|
|
|
| 5 |
A2 |
472 |
454 |
0.00 |
|
|
|
| 6 |
B1 |
491 |
472 |
108.91 |
|
|
|
| 7 |
B2 |
2119 |
2038 |
1232.06 |
|
|
|
| 8 |
B2 |
1259 |
1211 |
81.44 |
|
|
|
| 9 |
B2 |
465 |
447 |
11.37 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 4143.7 cm
-1
Scaled (by 0.9616) Zero Point Vibrational Energy (zpe) 3984.6 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at B3PW91/cc-pVTZ
Point Group is C2v
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| O1 |
0.000 |
0.000 |
0.492 |
| B2 |
0.000 |
1.252 |
0.062 |
| B3 |
0.000 |
-1.252 |
0.062 |
| O4 |
0.000 |
2.408 |
-0.285 |
| O5 |
0.000 |
-2.408 |
-0.285 |
Atom - Atom Distances (Å)
| |
O1 |
B2 |
B3 |
O4 |
O5 |
| O1 | | 1.3239 | 1.3239 | 2.5302 | 2.5302 |
B2 | 1.3239 | | 2.5039 | 1.2069 | 3.6763 | B3 | 1.3239 | 2.5039 | | 3.6763 | 1.2069 | O4 | 2.5302 | 1.2069 | 3.6763 | | 4.8161 | O5 | 2.5302 | 3.6763 | 1.2069 | 4.8161 | |
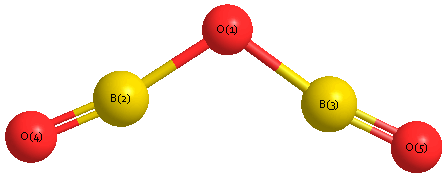 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| O1 |
B2 |
O4 |
177.698 |
|
O1 |
B3 |
O5 |
177.698 |
| B2 |
O1 |
B3 |
142.046 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B3PW91/cc-pVTZ
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
O |
0.006 |
|
|
|
| 2 |
B |
0.248 |
|
|
|
| 3 |
B |
0.248 |
|
|
|
| 4 |
O |
-0.251 |
|
|
|
| 5 |
O |
-0.251 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
1.185 |
1.185 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-24.341 |
0.000 |
0.000 |
| y |
0.000 |
-42.862 |
0.000 |
| z |
0.000 |
0.000 |
-25.035 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
9.608 |
0.000 |
0.000 |
| y |
0.000 |
-18.174 |
0.000 |
| z |
0.000 |
0.000 |
8.566 |
|
| Polar |
|---|
| 3z2-r2 | 17.132 |
|---|
| x2-y2 | 18.521 |
|---|
| xy | 0.000 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
3.236 |
0.000 |
0.000 |
| y |
0.000 |
6.831 |
0.000 |
| z |
0.000 |
0.000 |
3.433 |
<r2> (average value of r
2) Å
2
| <r2> |
130.928 |
| (<r2>)1/2 |
11.442 |
Jump to
S1C1
Energy calculated at B3PW91/cc-pVTZ
| | hartrees |
|---|
| Energy at 0K | -275.518100 |
| Energy at 298.15K | |
| HF Energy | -275.518100 |
| Nuclear repulsion energy | 117.103070 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at B3PW91/cc-pVTZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
Σg |
2121 |
2040 |
0.00 |
|
|
|
| 2 |
Σg |
679 |
653 |
0.00 |
|
|
|
| 3 |
Σu |
2151 |
2068 |
1569.09 |
|
|
|
| 4 |
Σu |
1332 |
1281 |
73.06 |
|
|
|
| 5 |
Πg |
474 |
456 |
0.00 |
|
|
|
| 5 |
Πg |
474 |
456 |
0.00 |
|
|
|
| 6 |
Πu |
460 |
443 |
117.29 |
|
|
|
| 6 |
Πu |
460 |
443 |
117.29 |
|
|
|
| 7 |
Πu |
85i |
82i |
5.32 |
|
|
|
| 7 |
Πu |
85i |
82i |
5.32 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 3990.9 cm
-1
Scaled (by 0.9616) Zero Point Vibrational Energy (zpe) 3837.6 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at B3PW91/cc-pVTZ
Point Group is D∞h
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| O1 |
0.000 |
0.000 |
0.000 |
| B2 |
0.000 |
0.000 |
1.310 |
| B3 |
0.000 |
0.000 |
-1.310 |
| O4 |
0.000 |
0.000 |
2.518 |
| O5 |
0.000 |
0.000 |
-2.518 |
Atom - Atom Distances (Å)
| |
O1 |
B2 |
B3 |
O4 |
O5 |
| O1 | | 1.3098 | 1.3098 | 2.5178 | 2.5178 |
B2 | 1.3098 | | 2.6195 | 1.2081 | 3.8276 | B3 | 1.3098 | 2.6195 | | 3.8276 | 1.2081 | O4 | 2.5178 | 1.2081 | 3.8276 | | 5.0357 | O5 | 2.5178 | 3.8276 | 1.2081 | 5.0357 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| O1 |
B2 |
O4 |
180.000 |
|
O1 |
B3 |
O5 |
180.000 |
| B2 |
O1 |
B3 |
180.000 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B3PW91/cc-pVTZ
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
O |
0.069 |
|
|
|
| 2 |
B |
0.248 |
|
|
|
| 3 |
B |
0.248 |
|
|
|
| 4 |
O |
-0.282 |
|
|
|
| 5 |
O |
-0.282 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
0.000 |
0.000 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-24.299 |
0.000 |
0.000 |
| y |
0.000 |
-24.299 |
0.000 |
| z |
0.000 |
0.000 |
-44.899 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
10.300 |
0.000 |
0.000 |
| y |
0.000 |
10.300 |
0.000 |
| z |
0.000 |
0.000 |
-20.600 |
|
| Polar |
|---|
| 3z2-r2 | -41.200 |
|---|
| x2-y2 | 0.000 |
|---|
| xy | 0.000 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
3.215 |
0.000 |
0.000 |
| y |
0.000 |
3.215 |
0.000 |
| z |
0.000 |
0.000 |
7.392 |
<r2> (average value of r
2) Å
2
| <r2> |
138.052 |
| (<r2>)1/2 |
11.750 |