Jump to
S1C2
Energy calculated at B3PW91/6-311G*
| | hartrees |
|---|
| Energy at 0K | -358.416925 |
| Energy at 298.15K | -358.422045 |
| Nuclear repulsion energy | 232.850084 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at B3PW91/6-311G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3755 |
3615 |
66.45 |
|
|
|
| 2 |
A' |
3750 |
3610 |
51.15 |
|
|
|
| 3 |
A' |
3619 |
3484 |
51.42 |
|
|
|
| 4 |
A' |
1845 |
1776 |
72.16 |
|
|
|
| 5 |
A' |
1825 |
1757 |
547.34 |
|
|
|
| 6 |
A' |
1636 |
1575 |
131.09 |
|
|
|
| 7 |
A' |
1433 |
1380 |
11.63 |
|
|
|
| 8 |
A' |
1333 |
1283 |
50.59 |
|
|
|
| 9 |
A' |
1212 |
1167 |
313.41 |
|
|
|
| 10 |
A' |
1109 |
1068 |
1.99 |
|
|
|
| 11 |
A' |
784 |
754 |
10.59 |
|
|
|
| 12 |
A' |
616 |
593 |
75.11 |
|
|
|
| 13 |
A' |
531 |
511 |
0.44 |
|
|
|
| 14 |
A' |
416 |
401 |
4.29 |
|
|
|
| 15 |
A' |
269 |
259 |
14.59 |
|
|
|
| 16 |
A" |
846 |
814 |
5.19 |
|
|
|
| 17 |
A" |
705 |
679 |
164.63 |
|
|
|
| 18 |
A" |
648 |
624 |
11.41 |
|
|
|
| 19 |
A" |
446 |
430 |
36.10 |
|
|
|
| 20 |
A" |
365 |
351 |
222.44 |
|
|
|
| 21 |
A" |
73 |
70 |
4.13 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 13608.5 cm
-1
Scaled (by 0.9627) Zero Point Vibrational Energy (zpe) 13100.9 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at B3PW91/6-311G*
Point Group is Cs
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.000 |
0.752 |
0.000 |
| C2 |
-0.057 |
-0.789 |
0.000 |
| O3 |
-1.094 |
-1.410 |
0.000 |
| O4 |
1.032 |
1.377 |
0.000 |
| O5 |
-1.211 |
1.297 |
0.000 |
| N6 |
1.189 |
-1.307 |
0.000 |
| H7 |
1.309 |
-2.305 |
0.000 |
| H8 |
1.990 |
-0.698 |
0.000 |
| H9 |
-1.093 |
2.258 |
0.000 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
O3 |
O4 |
O5 |
N6 |
H7 |
H8 |
H9 |
| C1 | | 1.5421 | 2.4227 | 1.2063 | 1.3278 | 2.3780 | 3.3261 | 2.4625 | 1.8608 |
C2 | 1.5421 | | 1.2082 | 2.4245 | 2.3833 | 1.3502 | 2.0415 | 2.0498 | 3.2181 | O3 | 2.4227 | 1.2082 | | 3.5049 | 2.7087 | 2.2857 | 2.5649 | 3.1654 | 3.6675 | O4 | 1.2063 | 2.4245 | 3.5049 | | 2.2441 | 2.6889 | 3.6931 | 2.2859 | 2.3001 | O5 | 1.3278 | 2.3833 | 2.7087 | 2.2441 | | 3.5413 | 4.3962 | 3.7718 | 0.9686 | N6 | 2.3780 | 1.3502 | 2.2857 | 2.6889 | 3.5413 | | 1.0055 | 1.0062 | 4.2331 | H7 | 3.3261 | 2.0415 | 2.5649 | 3.6931 | 4.3962 | 1.0055 | | 1.7457 | 5.1572 | H8 | 2.4625 | 2.0498 | 3.1654 | 2.2859 | 3.7718 | 1.0062 | 1.7457 | | 4.2715 | H9 | 1.8608 | 3.2181 | 3.6675 | 2.3001 | 0.9686 | 4.2331 | 5.1572 | 4.2715 | |
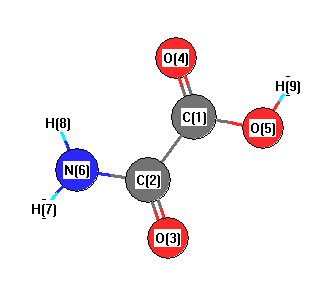 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
O3 |
123.044 |
|
C1 |
C2 |
N6 |
110.432 |
| C1 |
O5 |
H9 |
107.221 |
|
C2 |
C1 |
O4 |
123.349 |
| C2 |
C1 |
O5 |
112.076 |
|
C2 |
N6 |
H7 |
119.418 |
| C2 |
N6 |
H8 |
120.181 |
|
O3 |
C2 |
N6 |
126.524 |
| O4 |
C1 |
O5 |
124.574 |
|
H7 |
N6 |
H8 |
120.401 |
Electronic energy levels
Electronic state
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B3PW91/6-311G*
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
0.371 |
|
|
|
| 2 |
C |
0.408 |
|
|
|
| 3 |
O |
-0.337 |
|
|
|
| 4 |
O |
-0.347 |
|
|
|
| 5 |
O |
-0.474 |
|
|
|
| 6 |
N |
-0.796 |
|
|
|
| 7 |
H |
0.374 |
|
|
|
| 8 |
H |
0.390 |
|
|
|
| 9 |
H |
0.410 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
2.239 |
0.723 |
0.000 |
2.353 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-23.965 |
7.937 |
0.006 |
| y |
7.937 |
-38.805 |
0.002 |
| z |
0.006 |
0.002 |
-33.348 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
12.111 |
7.937 |
0.006 |
| y |
7.937 |
-10.148 |
0.002 |
| z |
0.006 |
0.002 |
-1.963 |
|
| Polar |
|---|
| 3z2-r2 | -3.927 |
|---|
| x2-y2 | 14.839 |
|---|
| xy | 7.937 |
|---|
| xz | 0.006 |
|---|
| yz | 0.002 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
0.000 |
0.000 |
0.000 |
| y |
0.000 |
0.000 |
0.000 |
| z |
0.000 |
0.000 |
0.000 |
<r2> (average value of r
2) Å
2
| <r2> |
141.131 |
| (<r2>)1/2 |
11.880 |
Jump to
S1C1
Energy calculated at B3PW91/6-311G*
| | hartrees |
|---|
| Energy at 0K | -358.422918 |
| Energy at 298.15K | -358.428271 |
| Nuclear repulsion energy | 234.344427 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at B3PW91/6-311G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3745 |
3605 |
71.22 |
|
|
|
| 2 |
A' |
3611 |
3477 |
62.70 |
|
|
|
| 3 |
A' |
3581 |
3448 |
111.87 |
|
|
|
| 4 |
A' |
1882 |
1812 |
235.09 |
|
|
|
| 5 |
A' |
1813 |
1745 |
338.10 |
|
|
|
| 6 |
A' |
1638 |
1577 |
78.17 |
|
|
|
| 7 |
A' |
1461 |
1407 |
240.71 |
|
|
|
| 8 |
A' |
1363 |
1312 |
353.31 |
|
|
|
| 9 |
A' |
1219 |
1174 |
9.72 |
|
|
|
| 10 |
A' |
1115 |
1073 |
3.68 |
|
|
|
| 11 |
A' |
811 |
781 |
10.66 |
|
|
|
| 12 |
A' |
635 |
612 |
12.93 |
|
|
|
| 13 |
A' |
548 |
527 |
2.02 |
|
|
|
| 14 |
A' |
410 |
395 |
8.29 |
|
|
|
| 15 |
A' |
276 |
266 |
40.88 |
|
|
|
| 16 |
A" |
835 |
804 |
2.20 |
|
|
|
| 17 |
A" |
769 |
741 |
124.77 |
|
|
|
| 18 |
A" |
676 |
651 |
3.64 |
|
|
|
| 19 |
A" |
485 |
467 |
195.31 |
|
|
|
| 20 |
A" |
406 |
391 |
92.90 |
|
|
|
| 21 |
A" |
120 |
116 |
6.56 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 13700.1 cm
-1
Scaled (by 0.9627) Zero Point Vibrational Energy (zpe) 13189.1 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at B3PW91/6-311G*
Point Group is Cs
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.010 |
-0.796 |
0.000 |
| C2 |
0.000 |
0.747 |
0.000 |
| O3 |
-1.067 |
1.339 |
0.000 |
| O4 |
1.016 |
-1.450 |
0.000 |
| O5 |
-1.221 |
-1.282 |
0.000 |
| N6 |
1.225 |
1.283 |
0.000 |
| H7 |
1.345 |
2.282 |
0.000 |
| H8 |
2.026 |
0.673 |
0.000 |
| H9 |
-1.820 |
-0.509 |
0.000 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
O3 |
O4 |
O5 |
N6 |
H7 |
H8 |
H9 |
| C1 | | 1.5429 | 2.3912 | 1.1999 | 1.3239 | 2.4078 | 3.3548 | 2.4940 | 1.8524 |
C2 | 1.5429 | | 1.2203 | 2.4209 | 2.3682 | 1.3370 | 2.0409 | 2.0274 | 2.2116 | O3 | 2.3912 | 1.2203 | | 3.4813 | 2.6254 | 2.2926 | 2.5900 | 3.1641 | 1.9958 | O4 | 1.1999 | 2.4209 | 3.4813 | | 2.2433 | 2.7417 | 3.7470 | 2.3513 | 2.9877 | O5 | 1.3239 | 2.3682 | 2.6254 | 2.2433 | | 3.5444 | 4.3918 | 3.7902 | 0.9773 | N6 | 2.4078 | 1.3370 | 2.2926 | 2.7417 | 3.5444 | | 1.0060 | 1.0074 | 3.5333 | H7 | 3.3548 | 2.0409 | 2.5900 | 3.7470 | 4.3918 | 1.0060 | | 1.7475 | 4.2201 | H8 | 2.4940 | 2.0274 | 3.1641 | 2.3513 | 3.7902 | 1.0074 | 1.7475 | | 4.0236 | H9 | 1.8524 | 2.2116 | 1.9958 | 2.9877 | 0.9773 | 3.5333 | 4.2201 | 4.0236 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
O3 |
119.391 |
|
C1 |
C2 |
N6 |
113.266 |
| C1 |
O5 |
H9 |
106.235 |
|
C2 |
C1 |
O4 |
123.445 |
| C2 |
C1 |
O5 |
111.166 |
|
C2 |
N6 |
H7 |
120.508 |
| C2 |
N6 |
H8 |
119.053 |
|
O3 |
C2 |
N6 |
127.343 |
| O4 |
C1 |
O5 |
125.390 |
|
H7 |
N6 |
H8 |
120.439 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B3PW91/6-311G*
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
0.416 |
|
|
|
| 2 |
C |
0.369 |
|
|
|
| 3 |
O |
-0.387 |
|
|
|
| 4 |
O |
-0.334 |
|
|
|
| 5 |
O |
-0.483 |
|
|
|
| 6 |
N |
-0.787 |
|
|
|
| 7 |
H |
0.378 |
|
|
|
| 8 |
H |
0.398 |
|
|
|
| 9 |
H |
0.429 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
1.550 |
2.710 |
0.000 |
3.122 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-29.950 |
7.229 |
0.000 |
| y |
7.229 |
-37.380 |
0.000 |
| z |
0.000 |
0.000 |
-33.294 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
5.387 |
7.229 |
0.000 |
| y |
7.229 |
-5.758 |
0.000 |
| z |
0.000 |
0.000 |
0.371 |
|
| Polar |
|---|
| 3z2-r2 | 0.741 |
|---|
| x2-y2 | 7.430 |
|---|
| xy | 7.229 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
6.972 |
0.009 |
0.000 |
| y |
0.009 |
5.515 |
0.000 |
| z |
0.000 |
0.000 |
2.760 |
<r2> (average value of r
2) Å
2
| <r2> |
138.886 |
| (<r2>)1/2 |
11.785 |