Vibrational Frequencies calculated at B3PW91/6-311G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3597 |
3463 |
0.15 |
|
|
|
| 2 |
A |
3579 |
3445 |
0.59 |
|
|
|
| 3 |
A |
3506 |
3375 |
3.79 |
|
|
|
| 4 |
A |
3504 |
3373 |
2.34 |
|
|
|
| 5 |
A |
3117 |
3001 |
37.21 |
|
|
|
| 6 |
A |
3100 |
2985 |
38.72 |
|
|
|
| 7 |
A |
3084 |
2969 |
29.85 |
|
|
|
| 8 |
A |
3037 |
2924 |
54.06 |
|
|
|
| 9 |
A |
3031 |
2918 |
18.88 |
|
|
|
| 10 |
A |
2908 |
2800 |
92.74 |
|
|
|
| 11 |
A |
1715 |
1651 |
11.90 |
|
|
|
| 12 |
A |
1703 |
1640 |
69.14 |
|
|
|
| 13 |
A |
1514 |
1458 |
4.92 |
|
|
|
| 14 |
A |
1503 |
1447 |
7.13 |
|
|
|
| 15 |
A |
1481 |
1425 |
1.44 |
|
|
|
| 16 |
A |
1425 |
1372 |
2.25 |
|
|
|
| 17 |
A |
1413 |
1361 |
12.35 |
|
|
|
| 18 |
A |
1407 |
1354 |
4.64 |
|
|
|
| 19 |
A |
1382 |
1330 |
11.29 |
|
|
|
| 20 |
A |
1347 |
1297 |
7.13 |
|
|
|
| 21 |
A |
1274 |
1227 |
1.17 |
|
|
|
| 22 |
A |
1217 |
1171 |
4.75 |
|
|
|
| 23 |
A |
1175 |
1131 |
5.17 |
|
|
|
| 24 |
A |
1123 |
1081 |
13.02 |
|
|
|
| 25 |
A |
1059 |
1019 |
13.43 |
|
|
|
| 26 |
A |
1029 |
990 |
0.96 |
|
|
|
| 27 |
A |
961 |
926 |
11.42 |
|
|
|
| 28 |
A |
912 |
878 |
88.37 |
|
|
|
| 29 |
A |
886 |
853 |
98.42 |
|
|
|
| 30 |
A |
845 |
814 |
95.71 |
|
|
|
| 31 |
A |
796 |
767 |
91.60 |
|
|
|
| 32 |
A |
494 |
475 |
2.98 |
|
|
|
| 33 |
A |
469 |
451 |
23.83 |
|
|
|
| 34 |
A |
373 |
359 |
57.04 |
|
|
|
| 35 |
A |
366 |
352 |
1.09 |
|
|
|
| 36 |
A |
255 |
246 |
6.01 |
|
|
|
| 37 |
A |
240 |
231 |
24.92 |
|
|
|
| 38 |
A |
218 |
210 |
38.23 |
|
|
|
| 39 |
A |
121 |
116 |
7.15 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 30582.0 cm
-1
Scaled (by 0.9627) Zero Point Vibrational Energy (zpe) 29441.3 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
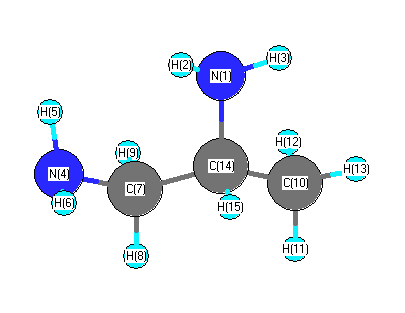
Charges from optimized geometry at B3PW91/6-311G*
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
N |
-0.762 |
|
|
|
| 2 |
H |
0.306 |
|
|
|
| 3 |
H |
0.326 |
|
|
|
| 4 |
N |
-0.755 |
|
|
|
| 5 |
H |
0.321 |
|
|
|
| 6 |
H |
0.310 |
|
|
|
| 7 |
C |
-0.249 |
|
|
|
| 8 |
H |
0.214 |
|
|
|
| 9 |
H |
0.222 |
|
|
|
| 10 |
C |
-0.651 |
|
|
|
| 11 |
H |
0.220 |
|
|
|
| 12 |
H |
0.229 |
|
|
|
| 13 |
H |
0.215 |
|
|
|
| 14 |
C |
-0.124 |
|
|
|
| 15 |
H |
0.177 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.985 |
1.201 |
1.769 |
2.354 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
0.000 |
0.000 |
0.000 |
| y |
0.000 |
0.000 |
0.000 |
| z |
0.000 |
0.000 |
0.000 |
<r2> (average value of r
2) Å
2
| <r2> |
143.559 |
| (<r2>)1/2 |
11.982 |