Vibrational Frequencies calculated at B3PW91/3-21G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3530 |
3387 |
0.41 |
|
|
|
| 2 |
A |
3526 |
3383 |
0.87 |
|
|
|
| 3 |
A |
3148 |
3020 |
37.11 |
|
|
|
| 4 |
A |
3138 |
3011 |
8.56 |
|
|
|
| 5 |
A |
3120 |
2993 |
25.70 |
|
|
|
| 6 |
A |
3087 |
2961 |
19.24 |
|
|
|
| 7 |
A |
3078 |
2953 |
16.94 |
|
|
|
| 8 |
A |
3051 |
2927 |
18.45 |
|
|
|
| 9 |
A |
2990 |
2868 |
93.49 |
|
|
|
| 10 |
A |
2981 |
2860 |
23.50 |
|
|
|
| 11 |
A |
1590 |
1525 |
3.52 |
|
|
|
| 12 |
A |
1557 |
1494 |
5.97 |
|
|
|
| 13 |
A |
1553 |
1490 |
9.24 |
|
|
|
| 14 |
A |
1533 |
1471 |
3.26 |
|
|
|
| 15 |
A |
1469 |
1409 |
0.43 |
|
|
|
| 16 |
A |
1451 |
1392 |
4.38 |
|
|
|
| 17 |
A |
1439 |
1380 |
27.51 |
|
|
|
| 18 |
A |
1396 |
1340 |
8.45 |
|
|
|
| 19 |
A |
1361 |
1306 |
12.36 |
|
|
|
| 20 |
A |
1351 |
1296 |
9.42 |
|
|
|
| 21 |
A |
1312 |
1259 |
31.89 |
|
|
|
| 22 |
A |
1269 |
1218 |
49.80 |
|
|
|
| 23 |
A |
1218 |
1168 |
5.19 |
|
|
|
| 24 |
A |
1161 |
1114 |
8.48 |
|
|
|
| 25 |
A |
1131 |
1085 |
49.63 |
|
|
|
| 26 |
A |
1096 |
1052 |
6.12 |
|
|
|
| 27 |
A |
1035 |
993 |
64.20 |
|
|
|
| 28 |
A |
1023 |
982 |
13.26 |
|
|
|
| 29 |
A |
1006 |
965 |
17.01 |
|
|
|
| 30 |
A |
955 |
916 |
15.17 |
|
|
|
| 31 |
A |
861 |
826 |
12.09 |
|
|
|
| 32 |
A |
826 |
793 |
9.66 |
|
|
|
| 33 |
A |
492 |
472 |
22.16 |
|
|
|
| 34 |
A |
463 |
444 |
1.78 |
|
|
|
| 35 |
A |
386 |
371 |
12.76 |
|
|
|
| 36 |
A |
335 |
322 |
7.12 |
|
|
|
| 37 |
A |
312 |
299 |
151.11 |
|
|
|
| 38 |
A |
278 |
267 |
115.39 |
|
|
|
| 39 |
A |
250 |
240 |
0.16 |
|
|
|
| 40 |
A |
183 |
176 |
7.02 |
|
|
|
| 41 |
A |
123 |
118 |
8.26 |
|
|
|
| 42 |
A |
101 |
97 |
7.92 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 31082.4 cm
-1
Scaled (by 0.9594) Zero Point Vibrational Energy (zpe) 29820.5 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
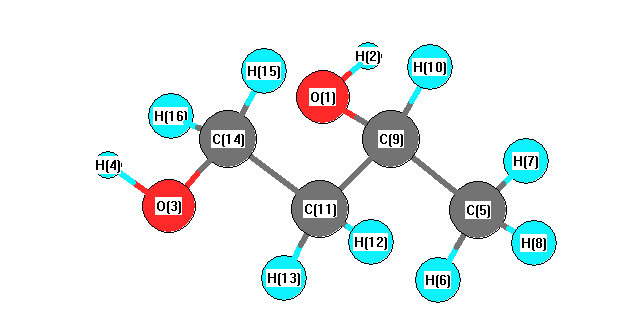
Charges from optimized geometry at B3PW91/3-21G*
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
O |
-0.560 |
|
|
|
| 2 |
H |
0.346 |
|
|
|
| 3 |
O |
-0.570 |
|
|
|
| 4 |
H |
0.345 |
|
|
|
| 5 |
C |
-0.624 |
|
|
|
| 6 |
H |
0.228 |
|
|
|
| 7 |
H |
0.198 |
|
|
|
| 8 |
H |
0.212 |
|
|
|
| 9 |
C |
-0.026 |
|
|
|
| 10 |
H |
0.195 |
|
|
|
| 11 |
C |
-0.423 |
|
|
|
| 12 |
H |
0.221 |
|
|
|
| 13 |
H |
0.236 |
|
|
|
| 14 |
C |
-0.174 |
|
|
|
| 15 |
H |
0.177 |
|
|
|
| 16 |
H |
0.220 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
1.382 |
1.076 |
1.464 |
2.283 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-30.931 |
-2.834 |
-0.216 |
| y |
-2.834 |
-37.783 |
0.892 |
| z |
-0.216 |
0.892 |
-38.290 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
7.105 |
-2.834 |
-0.216 |
| y |
-2.834 |
-3.173 |
0.892 |
| z |
-0.216 |
0.892 |
-3.933 |
|
| Polar |
|---|
| 3z2-r2 | -7.865 |
|---|
| x2-y2 | 6.852 |
|---|
| xy | -2.834 |
|---|
| xz | -0.216 |
|---|
| yz | 0.892 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
0.000 |
0.000 |
0.000 |
| y |
0.000 |
0.000 |
0.000 |
| z |
0.000 |
0.000 |
0.000 |
<r2> (average value of r
2) Å
2
| <r2> |
222.557 |
| (<r2>)1/2 |
14.918 |