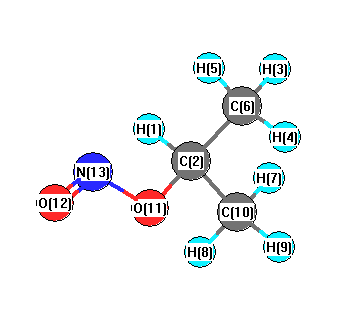
Vibrational Frequencies calculated at B3PW91/3-21G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3153 |
3025 |
22.47 |
|
|
|
| 2 |
A |
3148 |
3020 |
11.65 |
|
|
|
| 3 |
A |
3146 |
3019 |
27.98 |
|
|
|
| 4 |
A |
3141 |
3013 |
1.84 |
|
|
|
| 5 |
A |
3077 |
2952 |
5.57 |
|
|
|
| 6 |
A |
3068 |
2943 |
9.29 |
|
|
|
| 7 |
A |
3065 |
2940 |
11.78 |
|
|
|
| 8 |
A |
1568 |
1504 |
16.78 |
|
|
|
| 9 |
A |
1553 |
1490 |
11.17 |
|
|
|
| 10 |
A |
1545 |
1482 |
1.22 |
|
|
|
| 11 |
A |
1538 |
1476 |
1.79 |
|
|
|
| 12 |
A |
1479 |
1419 |
130.37 |
|
|
|
| 13 |
A |
1462 |
1403 |
6.10 |
|
|
|
| 14 |
A |
1445 |
1386 |
18.92 |
|
|
|
| 15 |
A |
1397 |
1341 |
9.82 |
|
|
|
| 16 |
A |
1349 |
1294 |
48.98 |
|
|
|
| 17 |
A |
1230 |
1180 |
24.28 |
|
|
|
| 18 |
A |
1171 |
1124 |
33.37 |
|
|
|
| 19 |
A |
1167 |
1120 |
13.94 |
|
|
|
| 20 |
A |
982 |
942 |
1.78 |
|
|
|
| 21 |
A |
964 |
925 |
0.39 |
|
|
|
| 22 |
A |
945 |
907 |
17.09 |
|
|
|
| 23 |
A |
886 |
850 |
164.47 |
|
|
|
| 24 |
A |
840 |
806 |
241.34 |
|
|
|
| 25 |
A |
562 |
539 |
4.04 |
|
|
|
| 26 |
A |
473 |
454 |
0.55 |
|
|
|
| 27 |
A |
403 |
387 |
5.33 |
|
|
|
| 28 |
A |
318 |
305 |
0.15 |
|
|
|
| 29 |
A |
275 |
264 |
0.96 |
|
|
|
| 30 |
A |
210 |
202 |
0.03 |
|
|
|
| 31 |
A |
207 |
199 |
0.84 |
|
|
|
| 32 |
A |
172 |
165 |
0.50 |
|
|
|
| 33 |
A |
42 |
40 |
0.16 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 22991.0 cm
-1
Scaled (by 0.9594) Zero Point Vibrational Energy (zpe) 22057.6 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B3PW91/3-21G*
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
H |
0.234 |
|
|
|
| 2 |
C |
-0.052 |
|
|
|
| 3 |
H |
0.213 |
|
|
|
| 4 |
H |
0.230 |
|
|
|
| 5 |
H |
0.227 |
|
|
|
| 6 |
C |
-0.607 |
|
|
|
| 7 |
H |
0.213 |
|
|
|
| 8 |
H |
0.227 |
|
|
|
| 9 |
H |
0.230 |
|
|
|
| 10 |
C |
-0.607 |
|
|
|
| 11 |
O |
-0.344 |
|
|
|
| 12 |
O |
-0.259 |
|
|
|
| 13 |
N |
0.296 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
2.413 |
0.000 |
-0.980 |
2.605 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-38.110 |
0.000 |
0.591 |
| y |
0.000 |
-34.327 |
0.000 |
| z |
0.591 |
0.000 |
-36.648 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-2.622 |
0.000 |
0.591 |
| y |
0.000 |
3.052 |
0.000 |
| z |
0.591 |
0.000 |
-0.430 |
|
| Polar |
|---|
| 3z2-r2 | -0.860 |
|---|
| x2-y2 | -3.783 |
|---|
| xy | 0.000 |
|---|
| xz | 0.591 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
0.000 |
0.000 |
0.000 |
| y |
0.000 |
0.000 |
0.000 |
| z |
0.000 |
0.000 |
0.000 |
<r2> (average value of r
2) Å
2
| <r2> |
197.877 |
| (<r2>)1/2 |
14.067 |