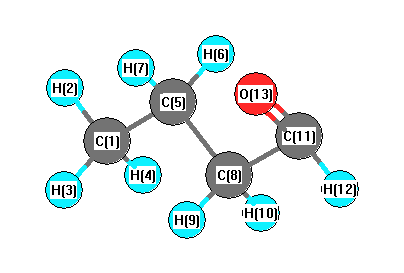
Vibrational Frequencies calculated at B3PW91/6-31G
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3138 |
3005 |
36.03 |
|
|
|
| 2 |
A' |
3069 |
2939 |
17.60 |
|
|
|
| 3 |
A' |
3046 |
2917 |
29.49 |
|
|
|
| 4 |
A' |
3019 |
2891 |
33.60 |
|
|
|
| 5 |
A' |
2956 |
2831 |
126.85 |
|
|
|
| 6 |
A' |
1745 |
1671 |
110.51 |
|
|
|
| 7 |
A' |
1548 |
1483 |
10.34 |
|
|
|
| 8 |
A' |
1535 |
1470 |
3.35 |
|
|
|
| 9 |
A' |
1492 |
1429 |
22.32 |
|
|
|
| 10 |
A' |
1461 |
1400 |
1.62 |
|
|
|
| 11 |
A' |
1442 |
1381 |
8.75 |
|
|
|
| 12 |
A' |
1417 |
1357 |
12.89 |
|
|
|
| 13 |
A' |
1346 |
1289 |
14.05 |
|
|
|
| 14 |
A' |
1164 |
1114 |
15.14 |
|
|
|
| 15 |
A' |
1081 |
1035 |
0.03 |
|
|
|
| 16 |
A' |
1002 |
960 |
5.95 |
|
|
|
| 17 |
A' |
882 |
844 |
8.02 |
|
|
|
| 18 |
A' |
702 |
673 |
13.67 |
|
|
|
| 19 |
A' |
355 |
340 |
0.78 |
|
|
|
| 20 |
A' |
200 |
191 |
9.81 |
|
|
|
| 21 |
A" |
3133 |
3000 |
61.22 |
|
|
|
| 22 |
A" |
3105 |
2974 |
0.26 |
|
|
|
| 23 |
A" |
3049 |
2920 |
8.33 |
|
|
|
| 24 |
A" |
1545 |
1480 |
9.96 |
|
|
|
| 25 |
A" |
1345 |
1288 |
0.21 |
|
|
|
| 26 |
A" |
1279 |
1225 |
0.41 |
|
|
|
| 27 |
A" |
1176 |
1126 |
0.02 |
|
|
|
| 28 |
A" |
972 |
931 |
0.06 |
|
|
|
| 29 |
A" |
817 |
782 |
1.62 |
|
|
|
| 30 |
A" |
699 |
670 |
7.57 |
|
|
|
| 31 |
A" |
246 |
236 |
0.03 |
|
|
|
| 32 |
A" |
175 |
167 |
1.61 |
|
|
|
| 33 |
A" |
76 |
73 |
1.98 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 25107.8 cm
-1
Scaled (by 0.9577) Zero Point Vibrational Energy (zpe) 24045.7 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B3PW91/6-31G
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.482 |
|
|
|
| 2 |
H |
0.166 |
|
|
|
| 3 |
H |
0.157 |
|
|
|
| 4 |
H |
0.157 |
|
|
|
| 5 |
C |
-0.294 |
|
|
|
| 6 |
H |
0.177 |
|
|
|
| 7 |
H |
0.177 |
|
|
|
| 8 |
C |
-0.393 |
|
|
|
| 9 |
H |
0.187 |
|
|
|
| 10 |
H |
0.187 |
|
|
|
| 11 |
C |
0.217 |
|
|
|
| 12 |
H |
0.136 |
|
|
|
| 13 |
O |
-0.392 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
1.951 |
2.113 |
0.000 |
2.876 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-36.849 |
-4.081 |
0.000 |
| y |
-4.081 |
-30.676 |
0.000 |
| z |
0.000 |
0.000 |
-30.574 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-6.224 |
-4.081 |
0.000 |
| y |
-4.081 |
3.035 |
0.000 |
| z |
0.000 |
0.000 |
3.189 |
|
| Polar |
|---|
| 3z2-r2 | 6.377 |
|---|
| x2-y2 | -6.173 |
|---|
| xy | -4.081 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
0.000 |
0.000 |
0.000 |
| y |
0.000 |
0.000 |
0.000 |
| z |
0.000 |
0.000 |
0.000 |
<r2> (average value of r
2) Å
2
| <r2> |
155.696 |
| (<r2>)1/2 |
12.478 |