Jump to
S1C2
S1C3
Energy calculated at MP2=FULL/STO-3G
| | hartrees |
|---|
| Energy at 0K | -251.181096 |
| Energy at 298.15K | -251.180833 |
| HF Energy | -250.974176 |
| Nuclear repulsion energy | 47.369306 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Geometric Data calculated at MP2=FULL/STO-3G
Point Group is C∞v
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.000 |
0.000 |
-0.593 |
| N2 |
0.000 |
0.000 |
-1.817 |
| Na3 |
0.000 |
0.000 |
1.480 |
Atom - Atom Distances (Å)
| |
C1 |
N2 |
Na3 |
| C1 | | 1.2238 | 2.0730 |
N2 | 1.2238 | | 3.2967 | Na3 | 2.0730 | 3.2967 | |
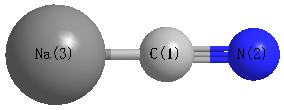 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
N2 |
Na3 |
0.000 |
|
C1 |
Na3 |
N2 |
0.000 |
| N2 |
C1 |
Na3 |
180.000 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Jump to
S1C1
S1C3
Energy calculated at MP2=FULL/STO-3G
| | hartrees |
|---|
| Energy at 0K | -251.172024 |
| Energy at 298.15K | -251.172011 |
| HF Energy | -250.976388 |
| Nuclear repulsion energy | 53.669122 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Geometric Data calculated at MP2=FULL/STO-3G
Point Group is Cs
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
1.065 |
0.447 |
0.000 |
| N2 |
0.000 |
1.080 |
0.000 |
| Na3 |
-0.581 |
-0.931 |
0.000 |
Atom - Atom Distances (Å)
| |
C1 |
N2 |
Na3 |
| C1 | | 1.2391 | 2.1472 |
N2 | 1.2391 | | 2.0931 | Na3 | 2.1472 | 2.0931 | |
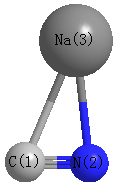 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
N2 |
Na3 |
75.418 |
|
C1 |
Na3 |
N2 |
33.952 |
| N2 |
C1 |
Na3 |
70.630 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Jump to
S1C1
S1C2
Energy calculated at MP2=FULL/STO-3G
| | hartrees |
|---|
| Energy at 0K | -251.166504 |
| Energy at 298.15K | -251.166082 |
| HF Energy | -250.978298 |
| Nuclear repulsion energy | 49.617731 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Geometric Data calculated at MP2=FULL/STO-3G
Point Group is C∞v
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.000 |
0.000 |
-1.826 |
| N2 |
0.000 |
0.000 |
-0.606 |
| Na3 |
0.000 |
0.000 |
1.381 |
Atom - Atom Distances (Å)
| |
C1 |
N2 |
Na3 |
| C1 | | 1.2198 | 3.2068 |
N2 | 1.2198 | | 1.9870 | Na3 | 3.2068 | 1.9870 | |
 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
N2 |
Na3 |
180.000 |
|
C1 |
Na3 |
N2 |
0.000 |
| N2 |
C1 |
Na3 |
0.000 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability