Jump to
S1C2
S2C1
S2C2
Energy calculated at MP2=FULL/STO-3G
| | hartrees |
|---|
| Energy at 0K | -129.141572 |
| Energy at 298.15K | -129.141066 |
| HF Energy | -129.019483 |
| Nuclear repulsion energy | 46.339373 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at MP2=FULL/STO-3G
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3555 |
3160 |
10.52 |
|
|
|
| 2 |
A' |
1997 |
1775 |
65.73 |
|
|
|
| 3 |
A' |
1074 |
955 |
16.22 |
|
|
|
| 4 |
A' |
920 |
818 |
14.26 |
|
|
|
| 5 |
A' |
443 |
394 |
1.50 |
|
|
|
| 6 |
A" |
517 |
459 |
3.15 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 4252.8 cm
-1
Scaled (by 0.8889) Zero Point Vibrational Energy (zpe) 3780.3 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at MP2=FULL/STO-3G
Point Group is Cs
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
-0.354 |
-1.251 |
0.000 |
| C2 |
0.000 |
0.127 |
0.000 |
| N3 |
0.259 |
1.268 |
0.000 |
| H4 |
0.310 |
-2.127 |
0.000 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
N3 |
H4 |
| C1 | | 1.4223 | 2.5922 | 1.0985 |
C2 | 1.4223 | | 1.1702 | 2.2745 | N3 | 2.5922 | 1.1702 | | 3.3949 | H4 | 1.0985 | 2.2745 | 3.3949 | |
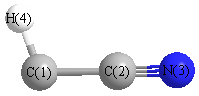 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
N3 |
178.384 |
|
C2 |
C1 |
H4 |
128.462 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Jump to
S1C1
S2C1
S2C2
Energy calculated at MP2=FULL/STO-3G
| | hartrees |
|---|
| Energy at 0K | -129.138863 |
| Energy at 298.15K | -129.138429 |
| HF Energy | -129.030038 |
| Nuclear repulsion energy | 45.993885 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at MP2=FULL/STO-3G
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
Σ |
3728 |
3314 |
105.64 |
|
|
|
| 2 |
Σ |
1636 |
1454 |
89.21 |
|
|
|
| 3 |
Σ |
767 |
682 |
160.87 |
|
|
|
| 4 |
Π |
721 |
641 |
13.10 |
|
|
|
| 4 |
Π |
721 |
641 |
13.10 |
|
|
|
| 5 |
Π |
438 |
390 |
0.60 |
|
|
|
| 5 |
Π |
438 |
390 |
0.60 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 4224.9 cm
-1
Scaled (by 0.8889) Zero Point Vibrational Energy (zpe) 3755.5 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at MP2=FULL/STO-3G
Point Group is C∞v
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.000 |
0.000 |
-1.212 |
| C2 |
0.000 |
0.000 |
0.004 |
| N3 |
0.000 |
0.000 |
1.363 |
| H4 |
0.000 |
0.000 |
-2.293 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
N3 |
H4 |
| C1 | | 1.2160 | 2.5744 | 1.0811 |
C2 | 1.2160 | | 1.3584 | 2.2971 | N3 | 2.5744 | 1.3584 | | 3.6555 | H4 | 1.0811 | 2.2971 | 3.6555 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
N3 |
180.000 |
|
C2 |
C1 |
H4 |
180.000 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at MP2=FULL/STO-3G
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.062 |
|
|
|
| 2 |
C |
-0.011 |
|
|
|
| 3 |
N |
-0.050 |
|
|
|
| 4 |
H |
0.123 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
-2.001 |
2.001 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-15.373 |
0.000 |
0.000 |
| y |
0.000 |
-15.373 |
0.000 |
| z |
0.000 |
0.000 |
-14.488 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-0.442 |
0.000 |
0.000 |
| y |
0.000 |
-0.442 |
0.000 |
| z |
0.000 |
0.000 |
0.885 |
|
| Polar |
|---|
| 3z2-r2 | 1.769 |
|---|
| x2-y2 | 0.000 |
|---|
| xy | 0.000 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
0.596 |
0.000 |
0.000 |
| y |
0.000 |
0.596 |
0.000 |
| z |
0.000 |
0.000 |
3.830 |
<r2> (average value of r
2) Å
2
| <r2> |
36.482 |
| (<r2>)1/2 |
6.040 |
Jump to
S1C1
S1C2
S2C2
Energy calculated at MP2=FULL/STO-3G
| | hartrees |
|---|
| Energy at 0K | -129.132140 |
| Energy at 298.15K | -129.131405 |
| HF Energy | -128.930149 |
| Nuclear repulsion energy | 44.434644 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at MP2=FULL/STO-3G
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3224 |
2866 |
24.78 |
|
|
|
| 2 |
A' |
2431 |
2161 |
67.33 |
|
|
|
| 3 |
A' |
1282 |
1139 |
17.96 |
|
|
|
| 4 |
A' |
891 |
792 |
26.27 |
|
|
|
| 5 |
A' |
342 |
304 |
6.89 |
|
|
|
| 6 |
A" |
347 |
309 |
6.59 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 4258.4 cm
-1
Scaled (by 0.8889) Zero Point Vibrational Energy (zpe) 3785.2 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at MP2=FULL/STO-3G
Point Group is Cs
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.125 |
-1.385 |
0.000 |
| C2 |
0.000 |
0.118 |
0.000 |
| N3 |
-0.287 |
1.305 |
0.000 |
| H4 |
1.260 |
-1.536 |
0.000 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
N3 |
H4 |
| C1 | | 1.5084 | 2.7219 | 1.1452 |
C2 | 1.5084 | | 1.2215 | 2.0796 | N3 | 2.7219 | 1.2215 | | 3.2355 | H4 | 1.1452 | 2.0796 | 3.2355 | |
More geometry information
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Jump to
S1C1
S1C2
S2C1
Energy calculated at MP2=FULL/STO-3G
| | hartrees |
|---|
| Energy at 0K | -129.132140 |
| Energy at 298.15K | -129.131405 |
| HF Energy | -128.930149 |
| Nuclear repulsion energy | 44.434644 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at MP2=FULL/STO-3G
Geometric Data calculated at MP2=FULL/STO-3G
Point Group is Cs
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability