Jump to
S1C2
Vibrational Frequencies calculated at MP2=FULL/STO-3G
Geometric Data calculated at MP2=FULL/STO-3G
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Jump to
S1C1
Energy calculated at MP2=FULL/STO-3G
| | hartrees |
|---|
| Energy at 0K | -352.107895 |
| Energy at 298.15K | |
| Nuclear repulsion energy | 225.480645 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at MP2=FULL/STO-3G
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
4033 |
3585 |
16.61 |
|
|
|
| 2 |
A' |
3808 |
3385 |
73.51 |
|
|
|
| 3 |
A' |
3589 |
3190 |
59.39 |
|
|
|
| 4 |
A' |
1889 |
1679 |
43.06 |
|
|
|
| 5 |
A' |
1861 |
1654 |
42.91 |
|
|
|
| 6 |
A' |
1727 |
1535 |
22.64 |
|
|
|
| 7 |
A' |
1635 |
1453 |
230.21 |
|
|
|
| 8 |
A' |
1403 |
1247 |
105.84 |
|
|
|
| 9 |
A' |
1217 |
1082 |
55.96 |
|
|
|
| 10 |
A' |
1111 |
988 |
6.69 |
|
|
|
| 11 |
A' |
789 |
701 |
5.22 |
|
|
|
| 12 |
A' |
622 |
553 |
8.30 |
|
|
|
| 13 |
A' |
540 |
480 |
5.93 |
|
|
|
| 14 |
A' |
365 |
324 |
16.40 |
|
|
|
| 15 |
A' |
197 |
175 |
21.75 |
|
|
|
| 16 |
A" |
901 |
801 |
66.18 |
|
|
|
| 17 |
A" |
734 |
652 |
3.90 |
|
|
|
| 18 |
A" |
570 |
507 |
1.47 |
|
|
|
| 19 |
A" |
387 |
344 |
5.29 |
|
|
|
| 20 |
A" |
141 |
125 |
4.86 |
|
|
|
| 21 |
A" |
503i |
448i |
274.22 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 13506.5 cm
-1
Scaled (by 0.8889) Zero Point Vibrational Energy (zpe) 12005.9 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
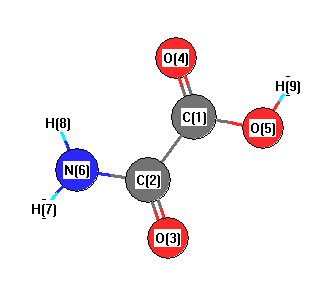
Geometric Data calculated at MP2=FULL/STO-3G
Point Group is Cs
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.061 |
-0.831 |
0.000 |
| C2 |
0.000 |
0.762 |
0.000 |
| O3 |
-1.161 |
1.281 |
0.000 |
| O4 |
1.089 |
-1.557 |
0.000 |
| O5 |
-1.261 |
-1.288 |
0.000 |
| N6 |
1.232 |
1.424 |
0.000 |
| H7 |
1.303 |
2.456 |
0.000 |
| H8 |
2.105 |
0.872 |
0.000 |
| H9 |
-1.732 |
-0.369 |
0.000 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
O3 |
O4 |
O5 |
N6 |
H7 |
H8 |
H9 |
| C1 | | 1.5944 | 2.4404 | 1.2582 | 1.3991 | 2.5415 | 3.5139 | 2.6608 | 1.8514 |
C2 | 1.5944 | | 1.2719 | 2.5616 | 2.4069 | 1.3986 | 2.1369 | 2.1081 | 2.0682 | O3 | 2.4404 | 1.2719 | | 3.6215 | 2.5709 | 2.3973 | 2.7297 | 3.2920 | 1.7456 | O4 | 1.2582 | 2.5616 | 3.6215 | | 2.3659 | 2.9843 | 4.0180 | 2.6324 | 3.0607 | O5 | 1.3991 | 2.4069 | 2.5709 | 2.3659 | | 3.6842 | 4.5377 | 3.9999 | 1.0324 | N6 | 2.5415 | 1.3986 | 2.3973 | 2.9843 | 3.6842 | | 1.0338 | 1.0336 | 3.4638 | H7 | 3.5139 | 2.1369 | 2.7297 | 4.0180 | 4.5377 | 1.0338 | | 1.7756 | 4.1457 | H8 | 2.6608 | 2.1081 | 3.2920 | 2.6324 | 3.9999 | 1.0336 | 1.7756 | | 4.0325 | H9 | 1.8514 | 2.0682 | 1.7456 | 3.0607 | 1.0324 | 3.4638 | 4.1457 | 4.0325 | |
 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
O3 |
116.275 |
|
C1 |
C2 |
N6 |
116.079 |
| C1 |
O5 |
H9 |
98.046 |
|
C2 |
C1 |
O4 |
127.389 |
| C2 |
C1 |
O5 |
106.857 |
|
C2 |
N6 |
H7 |
122.210 |
| C2 |
N6 |
H8 |
119.409 |
|
O3 |
C2 |
N6 |
127.645 |
| O4 |
C1 |
O5 |
125.753 |
|
H7 |
N6 |
H8 |
118.381 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability