Jump to
S1C2
Vibrational Frequencies calculated at MP2=FULL/6-311G**
Geometric Data calculated at MP2=FULL/6-311G**
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Jump to
S1C1
Energy calculated at MP2=FULL/6-311G**
| | hartrees |
|---|
| Energy at 0K | -235.249345 |
| Energy at 298.15K | -235.262947 |
| Nuclear repulsion energy | 255.739680 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at MP2=FULL/6-311G**
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3181 |
3017 |
51.07 |
|
|
|
| 2 |
A' |
3157 |
2994 |
37.13 |
|
|
|
| 3 |
A' |
3153 |
2991 |
31.34 |
|
|
|
| 4 |
A' |
3148 |
2986 |
10.34 |
|
|
|
| 5 |
A' |
3114 |
2954 |
29.88 |
|
|
|
| 6 |
A' |
3088 |
2929 |
6.76 |
|
|
|
| 7 |
A' |
3061 |
2904 |
27.66 |
|
|
|
| 8 |
A' |
3056 |
2899 |
25.88 |
|
|
|
| 9 |
A' |
1529 |
1450 |
2.48 |
|
|
|
| 10 |
A' |
1522 |
1444 |
2.45 |
|
|
|
| 11 |
A' |
1511 |
1433 |
7.09 |
|
|
|
| 12 |
A' |
1496 |
1419 |
0.19 |
|
|
|
| 13 |
A' |
1427 |
1354 |
2.89 |
|
|
|
| 14 |
A' |
1414 |
1341 |
12.31 |
|
|
|
| 15 |
A' |
1338 |
1270 |
4.70 |
|
|
|
| 16 |
A' |
1327 |
1259 |
4.22 |
|
|
|
| 17 |
A' |
1253 |
1188 |
0.36 |
|
|
|
| 18 |
A' |
1222 |
1159 |
0.24 |
|
|
|
| 19 |
A' |
1036 |
983 |
0.55 |
|
|
|
| 20 |
A' |
1012 |
960 |
0.53 |
|
|
|
| 21 |
A' |
971 |
921 |
0.26 |
|
|
|
| 22 |
A' |
947 |
898 |
0.41 |
|
|
|
| 23 |
A' |
895 |
849 |
0.32 |
|
|
|
| 24 |
A' |
720 |
683 |
0.76 |
|
|
|
| 25 |
A' |
606 |
575 |
1.26 |
|
|
|
| 26 |
A' |
401 |
380 |
0.54 |
|
|
|
| 27 |
A' |
355 |
337 |
0.01 |
|
|
|
| 28 |
A' |
196 |
186 |
0.00 |
|
|
|
| 29 |
A" |
3153 |
2991 |
22.36 |
|
|
|
| 30 |
A" |
3148 |
2986 |
33.95 |
|
|
|
| 31 |
A" |
3143 |
2981 |
7.39 |
|
|
|
| 32 |
A" |
3084 |
2926 |
64.17 |
|
|
|
| 33 |
A" |
1522 |
1444 |
8.43 |
|
|
|
| 34 |
A" |
1499 |
1422 |
0.31 |
|
|
|
| 35 |
A" |
1490 |
1414 |
4.10 |
|
|
|
| 36 |
A" |
1289 |
1222 |
0.66 |
|
|
|
| 37 |
A" |
1266 |
1201 |
0.17 |
|
|
|
| 38 |
A" |
1236 |
1173 |
0.00 |
|
|
|
| 39 |
A" |
1198 |
1137 |
2.06 |
|
|
|
| 40 |
A" |
1098 |
1042 |
0.05 |
|
|
|
| 41 |
A" |
998 |
947 |
0.01 |
|
|
|
| 42 |
A" |
962 |
913 |
2.04 |
|
|
|
| 43 |
A" |
904 |
858 |
0.00 |
|
|
|
| 44 |
A" |
804 |
762 |
0.17 |
|
|
|
| 45 |
A" |
384 |
364 |
0.01 |
|
|
|
| 46 |
A" |
303 |
288 |
0.01 |
|
|
|
| 47 |
A" |
275 |
261 |
0.00 |
|
|
|
| 48 |
A" |
226 |
214 |
0.01 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 37057.5 cm
-1
Scaled (by 0.9486) Zero Point Vibrational Energy (zpe) 35152.8 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
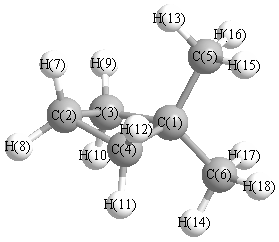
Geometric Data calculated at MP2=FULL/6-311G**
Point Group is Cs
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
-0.111 |
0.401 |
0.000 |
| C2 |
0.410 |
-1.705 |
0.000 |
| C3 |
-0.111 |
-0.721 |
1.075 |
| C4 |
-0.111 |
-0.721 |
-1.075 |
| C5 |
1.206 |
1.169 |
0.000 |
| C6 |
-1.294 |
1.352 |
0.000 |
| H7 |
1.501 |
-1.766 |
0.000 |
| H8 |
-0.001 |
-2.717 |
0.000 |
| H9 |
0.497 |
-0.559 |
1.970 |
| H10 |
0.497 |
-0.559 |
-1.970 |
| H11 |
-1.134 |
-0.971 |
1.374 |
| H12 |
-1.134 |
-0.971 |
-1.374 |
| H13 |
2.066 |
0.491 |
0.000 |
| H14 |
-2.238 |
0.797 |
0.000 |
| H15 |
1.278 |
1.807 |
-0.888 |
| H16 |
1.278 |
1.807 |
0.888 |
| H17 |
-1.276 |
1.996 |
0.887 |
| H18 |
-1.276 |
1.996 |
-0.887 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
C3 |
C4 |
C5 |
C6 |
H7 |
H8 |
H9 |
H10 |
H11 |
H12 |
H13 |
H14 |
H15 |
H16 |
H17 |
H18 |
| C1 | | 2.1692 | 1.5534 | 1.5534 | 1.5243 | 1.5181 | 2.7001 | 3.1200 | 2.2746 | 2.2746 | 2.1948 | 2.1948 | 2.1788 | 2.1636 | 2.1663 | 2.1663 | 2.1655 | 2.1655 |
C2 | 2.1692 | | 1.5475 | 1.5475 | 2.9819 | 3.4997 | 1.0924 | 1.0923 | 2.2811 | 2.2811 | 2.1930 | 2.1930 | 2.7509 | 3.6429 | 3.7249 | 3.7249 | 4.1625 | 4.1625 | C3 | 1.5534 | 1.5475 | | 2.1495 | 2.5414 | 2.6175 | 2.2008 | 2.2700 | 1.0947 | 3.1095 | 1.0951 | 2.6657 | 2.7135 | 2.8256 | 3.4883 | 2.8899 | 2.9622 | 3.5478 | C4 | 1.5534 | 1.5475 | 2.1495 | | 2.5414 | 2.6175 | 2.2008 | 2.2700 | 3.1095 | 1.0947 | 2.6657 | 1.0951 | 2.7135 | 2.8256 | 2.8899 | 3.4883 | 3.5478 | 2.9622 | C5 | 1.5243 | 2.9819 | 2.5414 | 2.5414 | | 2.5067 | 2.9492 | 4.0689 | 2.7147 | 2.7147 | 3.4558 | 3.4558 | 1.0947 | 3.4638 | 1.0958 | 1.0958 | 2.7627 | 2.7627 | C6 | 1.5181 | 3.4997 | 2.6175 | 2.6175 | 2.5067 | | 4.1869 | 4.2698 | 3.2778 | 3.2778 | 2.7038 | 2.7038 | 3.4686 | 1.0948 | 2.7581 | 2.7581 | 1.0962 | 1.0962 | H7 | 2.7001 | 1.0924 | 2.2008 | 2.2008 | 2.9492 | 4.1869 | | 1.7771 | 2.5190 | 2.5190 | 3.0757 | 3.0757 | 2.3271 | 4.5326 | 3.6882 | 3.6882 | 4.7590 | 4.7590 | H8 | 3.1200 | 1.0923 | 2.2700 | 2.2700 | 4.0689 | 4.2698 | 1.7771 | | 2.9645 | 2.9645 | 2.4942 | 2.4942 | 3.8166 | 4.1661 | 4.7843 | 4.7843 | 4.9627 | 4.9627 | H9 | 2.2746 | 2.2811 | 1.0947 | 3.1095 | 2.7147 | 3.2778 | 2.5190 | 2.9645 | | 3.9409 | 1.7853 | 3.7438 | 2.7289 | 3.6337 | 3.7916 | 2.7163 | 3.2936 | 4.2235 | H10 | 2.2746 | 2.2811 | 3.1095 | 1.0947 | 2.7147 | 3.2778 | 2.5190 | 2.9645 | 3.9409 | | 3.7438 | 1.7853 | 2.7289 | 3.6337 | 2.7163 | 3.7916 | 4.2235 | 3.2936 | H11 | 2.1948 | 2.1930 | 1.0951 | 2.6657 | 3.4558 | 2.7038 | 3.0757 | 2.4942 | 1.7853 | 3.7438 | | 2.7479 | 3.7773 | 2.4967 | 4.3184 | 3.7107 | 3.0102 | 3.7331 | H12 | 2.1948 | 2.1930 | 2.6657 | 1.0951 | 3.4558 | 2.7038 | 3.0757 | 2.4942 | 3.7438 | 1.7853 | 2.7479 | | 3.7773 | 2.4967 | 3.7107 | 4.3184 | 3.7331 | 3.0102 | H13 | 2.1788 | 2.7509 | 2.7135 | 2.7135 | 1.0947 | 3.4686 | 2.3271 | 3.8166 | 2.7289 | 2.7289 | 3.7773 | 3.7773 | | 4.3148 | 1.7723 | 1.7723 | 3.7711 | 3.7711 | H14 | 2.1636 | 3.6429 | 2.8256 | 2.8256 | 3.4638 | 1.0948 | 4.5326 | 4.1661 | 3.6337 | 3.6337 | 2.4967 | 2.4967 | 4.3148 | | 3.7636 | 3.7636 | 1.7744 | 1.7744 | H15 | 2.1663 | 3.7249 | 3.4883 | 2.8899 | 1.0958 | 2.7581 | 3.6882 | 4.7843 | 3.7916 | 2.7163 | 4.3184 | 3.7107 | 1.7723 | 3.7636 | | 1.7757 | 3.1155 | 2.5606 | H16 | 2.1663 | 3.7249 | 2.8899 | 3.4883 | 1.0958 | 2.7581 | 3.6882 | 4.7843 | 2.7163 | 3.7916 | 3.7107 | 4.3184 | 1.7723 | 3.7636 | 1.7757 | | 2.5606 | 3.1155 | H17 | 2.1655 | 4.1625 | 2.9622 | 3.5478 | 2.7627 | 1.0962 | 4.7590 | 4.9627 | 3.2936 | 4.2235 | 3.0102 | 3.7331 | 3.7711 | 1.7744 | 3.1155 | 2.5606 | | 1.7739 | H18 | 2.1655 | 4.1625 | 3.5478 | 2.9622 | 2.7627 | 1.0962 | 4.7590 | 4.9627 | 4.2235 | 3.2936 | 3.7331 | 3.0102 | 3.7711 | 1.7744 | 2.5606 | 3.1155 | 1.7739 | |
 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C3 |
C2 |
88.781 |
|
C1 |
C3 |
H9 |
117.351 |
| C1 |
C3 |
H11 |
110.738 |
|
C1 |
C4 |
C2 |
88.781 |
| C1 |
C4 |
H10 |
117.351 |
|
C1 |
C4 |
H12 |
110.738 |
| C1 |
C5 |
H13 |
111.541 |
|
C1 |
C5 |
H15 |
110.467 |
| C1 |
C5 |
H16 |
110.467 |
|
C1 |
C6 |
H14 |
110.756 |
| C1 |
C6 |
H17 |
110.821 |
|
C1 |
C6 |
H18 |
110.821 |
| C2 |
C3 |
H9 |
118.385 |
|
C2 |
C3 |
H11 |
111.016 |
| C2 |
C4 |
H10 |
118.385 |
|
C2 |
C4 |
H12 |
111.016 |
| C3 |
C1 |
C4 |
87.555 |
|
C3 |
C1 |
C5 |
111.325 |
| C3 |
C1 |
C6 |
116.899 |
|
C3 |
C2 |
C4 |
87.976 |
| C3 |
C2 |
H7 |
111.797 |
|
C3 |
C2 |
H8 |
117.582 |
| C4 |
C1 |
C5 |
111.325 |
|
C4 |
C1 |
C6 |
116.899 |
| C4 |
C2 |
H7 |
111.797 |
|
C4 |
C2 |
H8 |
117.582 |
| C5 |
C1 |
C6 |
110.958 |
|
H7 |
C2 |
H8 |
108.873 |
| H9 |
C3 |
H11 |
109.228 |
|
H10 |
C4 |
H12 |
109.228 |
| H13 |
C5 |
H15 |
108.010 |
|
H13 |
C5 |
H16 |
108.010 |
| H14 |
C6 |
H17 |
108.159 |
|
H14 |
C6 |
H18 |
108.159 |
| H15 |
C5 |
H16 |
108.232 |
|
H17 |
C6 |
H18 |
108.016 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability