Jump to
S1C2
Energy calculated at MP2=FULL/6-311G**
| | hartrees |
|---|
| Energy at 0K | -322.960521 |
| Energy at 298.15K | |
| HF Energy | -321.818974 |
| Nuclear repulsion energy | 234.511409 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at MP2=FULL/6-311G**
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3179 |
3016 |
23.64 |
|
|
|
| 2 |
A' |
3104 |
2944 |
25.70 |
|
|
|
| 3 |
A' |
3083 |
2925 |
37.10 |
|
|
|
| 4 |
A' |
3080 |
2922 |
8.73 |
|
|
|
| 5 |
A' |
1661 |
1575 |
127.08 |
|
|
|
| 6 |
A' |
1544 |
1464 |
16.79 |
|
|
|
| 7 |
A' |
1528 |
1449 |
3.22 |
|
|
|
| 8 |
A' |
1514 |
1436 |
0.96 |
|
|
|
| 9 |
A' |
1445 |
1371 |
21.07 |
|
|
|
| 10 |
A' |
1434 |
1360 |
7.07 |
|
|
|
| 11 |
A' |
1347 |
1277 |
6.91 |
|
|
|
| 12 |
A' |
1184 |
1123 |
6.74 |
|
|
|
| 13 |
A' |
1114 |
1056 |
38.77 |
|
|
|
| 14 |
A' |
1078 |
1023 |
7.72 |
|
|
|
| 15 |
A' |
945 |
897 |
86.48 |
|
|
|
| 16 |
A' |
848 |
805 |
492.23 |
|
|
|
| 17 |
A' |
666 |
632 |
88.40 |
|
|
|
| 18 |
A' |
395 |
375 |
0.50 |
|
|
|
| 19 |
A' |
354 |
336 |
3.95 |
|
|
|
| 20 |
A' |
157 |
149 |
0.38 |
|
|
|
| 21 |
A" |
3175 |
3012 |
56.42 |
|
|
|
| 22 |
A" |
3155 |
2993 |
2.66 |
|
|
|
| 23 |
A" |
3136 |
2975 |
9.35 |
|
|
|
| 24 |
A" |
1521 |
1443 |
8.06 |
|
|
|
| 25 |
A" |
1338 |
1269 |
0.10 |
|
|
|
| 26 |
A" |
1293 |
1227 |
0.42 |
|
|
|
| 27 |
A" |
1209 |
1147 |
1.19 |
|
|
|
| 28 |
A" |
912 |
865 |
1.27 |
|
|
|
| 29 |
A" |
781 |
741 |
1.38 |
|
|
|
| 30 |
A" |
243 |
231 |
0.03 |
|
|
|
| 31 |
A" |
212 |
201 |
0.91 |
|
|
|
| 32 |
A" |
99 |
94 |
1.32 |
|
|
|
| 33 |
A" |
55i |
52i |
0.14 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 23339.0 cm
-1
Scaled (by 0.9486) Zero Point Vibrational Energy (zpe) 22139.3 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at MP2=FULL/6-311G**
Point Group is Cs
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
-1.902 |
2.211 |
0.000 |
| C2 |
-1.506 |
0.736 |
0.000 |
| C3 |
0.000 |
0.572 |
0.000 |
| O4 |
0.277 |
-0.837 |
0.000 |
| N5 |
1.671 |
-1.032 |
0.000 |
| O6 |
1.936 |
-2.186 |
0.000 |
| H7 |
-2.988 |
2.324 |
0.000 |
| H8 |
-1.510 |
2.721 |
0.885 |
| H9 |
-1.510 |
2.721 |
-0.885 |
| H10 |
-1.914 |
0.232 |
-0.881 |
| H11 |
-1.914 |
0.232 |
0.881 |
| H12 |
0.440 |
1.033 |
0.891 |
| H13 |
0.440 |
1.033 |
-0.891 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
C3 |
O4 |
N5 |
O6 |
H7 |
H8 |
H9 |
H10 |
H11 |
H12 |
H13 |
| C1 | | 1.5269 | 2.5102 | 3.7461 | 4.8253 | 5.8357 | 1.0922 | 1.0939 | 1.0939 | 2.1662 | 2.1662 | 2.7680 | 2.7680 |
C2 | 1.5269 | | 1.5147 | 2.3773 | 3.6361 | 4.5146 | 2.1720 | 2.1736 | 2.1736 | 1.0940 | 1.0940 | 2.1603 | 2.1603 | C3 | 2.5102 | 1.5147 | | 1.4358 | 2.3168 | 3.3696 | 3.4635 | 2.7718 | 2.7718 | 2.1340 | 2.1340 | 1.0952 | 1.0952 | O4 | 3.7461 | 2.3773 | 1.4358 | | 1.4081 | 2.1382 | 4.5439 | 4.0788 | 4.0788 | 2.5917 | 2.5917 | 2.0777 | 2.0777 | N5 | 4.8253 | 3.6361 | 2.3168 | 1.4081 | | 1.1837 | 5.7421 | 4.9995 | 4.9995 | 3.9022 | 3.9022 | 2.5645 | 2.5645 | O6 | 5.8357 | 4.5146 | 3.3696 | 2.1382 | 1.1837 | | 6.6767 | 6.0613 | 6.0613 | 4.6303 | 4.6303 | 3.6599 | 3.6599 | H7 | 1.0922 | 2.1720 | 3.4635 | 4.5439 | 5.7421 | 6.6767 | | 1.7676 | 1.7676 | 2.5111 | 2.5111 | 3.7693 | 3.7693 | H8 | 1.0939 | 2.1736 | 2.7718 | 4.0788 | 4.9995 | 6.0613 | 1.7676 | | 1.7693 | 3.0787 | 2.5219 | 2.5792 | 3.1312 | H9 | 1.0939 | 2.1736 | 2.7718 | 4.0788 | 4.9995 | 6.0613 | 1.7676 | 1.7693 | | 2.5219 | 3.0787 | 3.1312 | 2.5792 | H10 | 2.1662 | 1.0940 | 2.1340 | 2.5917 | 3.9022 | 4.6303 | 2.5111 | 3.0787 | 2.5219 | | 1.7623 | 3.0529 | 2.4860 | H11 | 2.1662 | 1.0940 | 2.1340 | 2.5917 | 3.9022 | 4.6303 | 2.5111 | 2.5219 | 3.0787 | 1.7623 | | 2.4860 | 3.0529 | H12 | 2.7680 | 2.1603 | 1.0952 | 2.0777 | 2.5645 | 3.6599 | 3.7693 | 2.5792 | 3.1312 | 3.0529 | 2.4860 | | 1.7816 | H13 | 2.7680 | 2.1603 | 1.0952 | 2.0777 | 2.5645 | 3.6599 | 3.7693 | 3.1312 | 2.5792 | 2.4860 | 3.0529 | 1.7816 | |
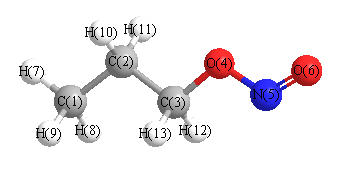 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
C3 |
111.234 |
|
C1 |
C2 |
H10 |
110.388 |
| C1 |
C2 |
H11 |
110.388 |
|
C2 |
C1 |
H7 |
110.956 |
| C2 |
C1 |
H8 |
110.986 |
|
C2 |
C1 |
H9 |
110.986 |
| C2 |
C3 |
O4 |
107.333 |
|
C2 |
C3 |
H12 |
110.705 |
| C2 |
C3 |
H13 |
110.705 |
|
C3 |
C2 |
H10 |
108.709 |
| C3 |
C2 |
H11 |
108.709 |
|
C3 |
O4 |
N5 |
109.103 |
| O4 |
C3 |
H12 |
109.613 |
|
O4 |
C3 |
H13 |
109.613 |
| O4 |
N5 |
O6 |
110.875 |
|
H7 |
C1 |
H8 |
107.913 |
| H7 |
C1 |
H9 |
107.913 |
|
H8 |
C1 |
H9 |
107.950 |
| H10 |
C2 |
H11 |
107.303 |
|
H12 |
C3 |
H13 |
108.852 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Jump to
S1C1
Energy calculated at MP2=FULL/6-311G**
| | hartrees |
|---|
| Energy at 0K | -322.962879 |
| Energy at 298.15K | -322.971974 |
| HF Energy | -321.818124 |
| Nuclear repulsion energy | 242.008672 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at MP2=FULL/6-311G**
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3184 |
3021 |
23.14 |
|
|
|
| 2 |
A |
3173 |
3010 |
32.85 |
|
|
|
| 3 |
A |
3157 |
2995 |
22.00 |
|
|
|
| 4 |
A |
3141 |
2979 |
11.15 |
|
|
|
| 5 |
A |
3091 |
2933 |
38.66 |
|
|
|
| 6 |
A |
3087 |
2928 |
10.98 |
|
|
|
| 7 |
A |
3085 |
2926 |
17.59 |
|
|
|
| 8 |
A |
1670 |
1584 |
149.80 |
|
|
|
| 9 |
A |
1529 |
1451 |
8.92 |
|
|
|
| 10 |
A |
1521 |
1443 |
7.61 |
|
|
|
| 11 |
A |
1510 |
1432 |
3.00 |
|
|
|
| 12 |
A |
1498 |
1421 |
2.65 |
|
|
|
| 13 |
A |
1438 |
1365 |
8.59 |
|
|
|
| 14 |
A |
1426 |
1353 |
11.28 |
|
|
|
| 15 |
A |
1395 |
1324 |
1.07 |
|
|
|
| 16 |
A |
1323 |
1255 |
5.42 |
|
|
|
| 17 |
A |
1312 |
1245 |
7.93 |
|
|
|
| 18 |
A |
1206 |
1144 |
12.18 |
|
|
|
| 19 |
A |
1139 |
1081 |
11.44 |
|
|
|
| 20 |
A |
1121 |
1063 |
32.11 |
|
|
|
| 21 |
A |
1026 |
973 |
10.84 |
|
|
|
| 22 |
A |
944 |
895 |
63.74 |
|
|
|
| 23 |
A |
904 |
858 |
1.52 |
|
|
|
| 24 |
A |
835 |
792 |
338.64 |
|
|
|
| 25 |
A |
789 |
749 |
105.73 |
|
|
|
| 26 |
A |
609 |
578 |
83.07 |
|
|
|
| 27 |
A |
458 |
434 |
18.77 |
|
|
|
| 28 |
A |
392 |
372 |
3.58 |
|
|
|
| 29 |
A |
284 |
270 |
2.01 |
|
|
|
| 30 |
A |
259 |
245 |
1.19 |
|
|
|
| 31 |
A |
185 |
175 |
0.42 |
|
|
|
| 32 |
A |
114 |
108 |
0.93 |
|
|
|
| 33 |
A |
56 |
53 |
0.06 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 23430.0 cm
-1
Scaled (by 0.9486) Zero Point Vibrational Energy (zpe) 22225.7 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at MP2=FULL/6-311G**
Point Group is C1
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
-2.158 |
-0.930 |
0.183 |
| C2 |
-1.757 |
0.458 |
-0.309 |
| C3 |
-0.409 |
0.899 |
0.221 |
| O4 |
0.560 |
-0.028 |
-0.299 |
| N5 |
1.778 |
0.210 |
0.315 |
| O6 |
2.568 |
-0.553 |
-0.140 |
| H7 |
-3.138 |
-1.214 |
-0.207 |
| H8 |
-2.208 |
-0.952 |
1.276 |
| H9 |
-1.429 |
-1.676 |
-0.140 |
| H10 |
-1.725 |
0.479 |
-1.403 |
| H11 |
-2.494 |
1.203 |
0.008 |
| H12 |
-0.154 |
1.909 |
-0.112 |
| H13 |
-0.385 |
0.862 |
1.315 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
C3 |
O4 |
N5 |
O6 |
H7 |
H8 |
H9 |
H10 |
H11 |
H12 |
H13 |
| C1 | | 1.5256 | 2.5300 | 2.9037 | 4.0996 | 4.7518 | 1.0929 | 1.0943 | 1.0916 | 2.1650 | 2.1661 | 3.4867 | 2.7628 |
C2 | 1.5256 | | 1.5138 | 2.3669 | 3.5980 | 4.4444 | 2.1711 | 2.1684 | 2.1652 | 1.0945 | 1.0958 | 2.1707 | 2.1642 | C3 | 2.5300 | 1.5138 | | 1.4381 | 2.2951 | 3.3319 | 3.4776 | 2.7874 | 2.7928 | 2.1326 | 2.1178 | 1.0940 | 1.0948 | O4 | 2.9037 | 2.3669 | 1.4381 | | 1.3849 | 2.0817 | 3.8851 | 3.3157 | 2.5885 | 2.5884 | 3.3072 | 2.0725 | 2.0712 | N5 | 4.0996 | 3.5980 | 2.2951 | 1.3849 | | 1.1888 | 5.1451 | 4.2613 | 3.7487 | 3.9118 | 4.3969 | 2.6084 | 2.4705 | O6 | 4.7518 | 4.4444 | 3.3319 | 2.0817 | 1.1888 | | 5.7451 | 4.9973 | 4.1524 | 4.5930 | 5.3602 | 3.6705 | 3.5831 | H7 | 1.0929 | 2.1711 | 3.4776 | 3.8851 | 5.1451 | 5.7451 | | 1.7699 | 1.7717 | 2.5089 | 2.5105 | 4.3202 | 3.7690 | H8 | 1.0943 | 2.1684 | 2.7874 | 3.3157 | 4.2613 | 4.9973 | 1.7699 | | 1.7703 | 3.0752 | 2.5162 | 3.7848 | 2.5714 | H9 | 1.0916 | 2.1652 | 2.7928 | 2.5885 | 3.7487 | 4.1524 | 1.7717 | 1.7703 | | 2.5159 | 3.0733 | 3.8050 | 3.1060 | H10 | 2.1650 | 1.0945 | 2.1326 | 2.5884 | 3.9118 | 4.5930 | 2.5089 | 3.0752 | 2.5159 | | 1.7627 | 2.4857 | 3.0551 | H11 | 2.1661 | 1.0958 | 2.1178 | 3.3072 | 4.3969 | 5.3602 | 2.5105 | 2.5162 | 3.0733 | 1.7627 | | 2.4468 | 2.5047 | H12 | 3.4867 | 2.1707 | 1.0940 | 2.0725 | 2.6084 | 3.6705 | 4.3202 | 3.7848 | 3.8050 | 2.4857 | 2.4468 | | 1.7854 | H13 | 2.7628 | 2.1642 | 1.0948 | 2.0712 | 2.4705 | 3.5831 | 3.7690 | 2.5714 | 3.1060 | 3.0551 | 2.5047 | 1.7854 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
C3 |
112.695 |
|
C1 |
C2 |
H10 |
110.360 |
| C1 |
C2 |
H11 |
110.370 |
|
C2 |
C1 |
H7 |
110.940 |
| C2 |
C1 |
H8 |
110.643 |
|
C2 |
C1 |
H9 |
110.550 |
| C2 |
C3 |
O4 |
106.580 |
|
C2 |
C3 |
H12 |
111.679 |
| C2 |
C3 |
H13 |
111.111 |
|
C3 |
C2 |
H10 |
108.624 |
| C3 |
C2 |
H11 |
107.415 |
|
C3 |
O4 |
N5 |
108.766 |
| O4 |
C3 |
H12 |
109.115 |
|
O4 |
C3 |
H13 |
108.958 |
| O4 |
N5 |
O6 |
107.723 |
|
H7 |
C1 |
H8 |
108.042 |
| H7 |
C1 |
H9 |
108.397 |
|
H8 |
C1 |
H9 |
108.171 |
| H10 |
C2 |
H11 |
107.177 |
|
H12 |
C3 |
H13 |
109.315 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability