Jump to
S1C2
Vibrational Frequencies calculated at MP2=FULL/TZVP
Geometric Data calculated at MP2=FULL/TZVP
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Jump to
S1C1
Energy calculated at MP2=FULL/TZVP
| | hartrees |
|---|
| Energy at 0K | -997.772310 |
| Energy at 298.15K | -997.777237 |
| HF Energy | -997.103559 |
| Nuclear repulsion energy | 202.535449 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at MP2=FULL/TZVP
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3207 |
3055 |
1.26 |
|
|
|
| 2 |
A |
3140 |
2992 |
23.28 |
|
|
|
| 3 |
A |
1491 |
1421 |
0.28 |
|
|
|
| 4 |
A |
1395 |
1329 |
22.30 |
|
|
|
| 5 |
A |
1262 |
1202 |
1.36 |
|
|
|
| 6 |
A |
1087 |
1036 |
0.72 |
|
|
|
| 7 |
A |
991 |
944 |
8.44 |
|
|
|
| 8 |
A |
701 |
667 |
16.93 |
|
|
|
| 9 |
A |
274 |
261 |
0.62 |
|
|
|
| 10 |
A |
125 |
119 |
0.84 |
|
|
|
| 11 |
B |
3218 |
3066 |
4.81 |
|
|
|
| 12 |
B |
3134 |
2986 |
4.09 |
|
|
|
| 13 |
B |
1486 |
1416 |
10.53 |
|
|
|
| 14 |
B |
1373 |
1308 |
42.34 |
|
|
|
| 15 |
B |
1197 |
1140 |
1.39 |
|
|
|
| 16 |
B |
927 |
883 |
15.31 |
|
|
|
| 17 |
B |
727 |
692 |
18.64 |
|
|
|
| 18 |
B |
423 |
403 |
7.08 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 13078.3 cm
-1
Scaled (by 0.9527) Zero Point Vibrational Energy (zpe) 12459.7 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at MP2=FULL/TZVP
Point Group is C2
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.295 |
0.693 |
0.894 |
| C2 |
-0.295 |
-0.693 |
0.894 |
| Cl3 |
-0.295 |
1.685 |
-0.471 |
| Cl4 |
0.295 |
-1.685 |
-0.471 |
| H5 |
0.003 |
1.201 |
1.810 |
| H6 |
1.378 |
0.654 |
0.827 |
| H7 |
-0.003 |
-1.201 |
1.810 |
| H8 |
-1.378 |
-0.654 |
0.827 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
Cl3 |
Cl4 |
H5 |
H6 |
H7 |
H8 |
| C1 | | 1.5061 | 1.7879 | 2.7422 | 1.0876 | 1.0856 | 2.1248 | 2.1485 |
C2 | 1.5061 | | 2.7422 | 1.7879 | 2.1248 | 2.1485 | 1.0876 | 1.0856 | Cl3 | 1.7879 | 2.7422 | | 3.4217 | 2.3510 | 2.3551 | 3.6906 | 2.8860 | Cl4 | 2.7422 | 1.7879 | 3.4217 | | 3.6906 | 2.8860 | 2.3510 | 2.3551 | H5 | 1.0876 | 2.1248 | 2.3510 | 3.6906 | | 1.7769 | 2.4021 | 2.5125 | H6 | 1.0856 | 2.1485 | 2.3551 | 2.8860 | 1.7769 | | 2.5125 | 3.0500 | H7 | 2.1248 | 1.0876 | 3.6906 | 2.3510 | 2.4021 | 2.5125 | | 1.7769 | H8 | 2.1485 | 1.0856 | 2.8860 | 2.3551 | 2.5125 | 3.0500 | 1.7769 | |
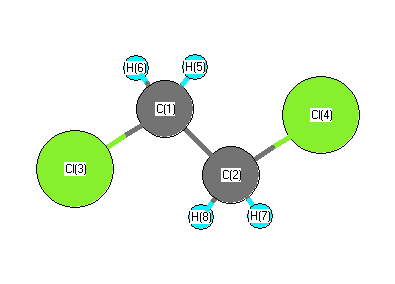 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
Cl4 |
112.427 |
|
C1 |
C2 |
H7 |
108.943 |
| C1 |
C2 |
H8 |
110.946 |
|
C2 |
C1 |
Cl3 |
112.427 |
| C2 |
C1 |
H5 |
108.943 |
|
C2 |
C1 |
H6 |
110.946 |
| Cl3 |
C1 |
H5 |
107.170 |
|
Cl3 |
C1 |
H6 |
107.563 |
| Cl4 |
C2 |
H7 |
107.170 |
|
Cl4 |
C2 |
H8 |
107.563 |
| H5 |
C1 |
H6 |
109.697 |
|
H7 |
C2 |
H8 |
109.697 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability