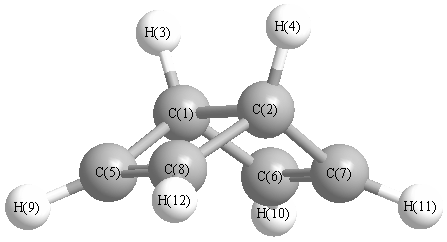
Vibrational Frequencies calculated at MP2=FULL/TZVP
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A1 |
3263 |
3109 |
7.31 |
294.72 |
0.03 |
0.06 |
| 2 |
A1 |
3165 |
3015 |
23.15 |
150.83 |
0.11 |
0.20 |
| 3 |
A1 |
1592 |
1516 |
0.14 |
23.04 |
0.01 |
0.02 |
| 4 |
A1 |
1182 |
1126 |
4.19 |
26.13 |
0.12 |
0.22 |
| 5 |
A1 |
1044 |
995 |
2.42 |
6.42 |
0.75 |
0.85 |
| 6 |
A1 |
967 |
921 |
6.87 |
9.20 |
0.22 |
0.37 |
| 7 |
A1 |
876 |
835 |
2.09 |
4.99 |
0.73 |
0.84 |
| 8 |
A1 |
827 |
788 |
77.96 |
6.30 |
0.03 |
0.05 |
| 9 |
A1 |
402 |
383 |
4.81 |
6.93 |
0.57 |
0.73 |
| 10 |
A2 |
3229 |
3077 |
0.00 |
152.90 |
0.75 |
0.86 |
| 11 |
A2 |
1300 |
1239 |
0.00 |
3.84 |
0.75 |
0.86 |
| 12 |
A2 |
1212 |
1155 |
0.00 |
10.06 |
0.75 |
0.86 |
| 13 |
A2 |
975 |
929 |
0.00 |
0.58 |
0.75 |
0.86 |
| 14 |
A2 |
882 |
840 |
0.00 |
0.89 |
0.75 |
0.86 |
| 15 |
A2 |
772 |
735 |
0.00 |
1.98 |
0.75 |
0.86 |
| 16 |
A2 |
305 |
290 |
0.00 |
1.67 |
0.75 |
0.86 |
| 17 |
B1 |
3261 |
3107 |
35.28 |
60.09 |
0.75 |
0.86 |
| 18 |
B1 |
1568 |
1494 |
9.15 |
0.69 |
0.75 |
0.86 |
| 19 |
B1 |
1223 |
1165 |
2.24 |
1.43 |
0.75 |
0.86 |
| 20 |
B1 |
1118 |
1065 |
0.45 |
0.02 |
0.75 |
0.86 |
| 21 |
B1 |
1026 |
977 |
0.10 |
6.55 |
0.75 |
0.86 |
| 22 |
B1 |
737 |
702 |
50.17 |
2.74 |
0.75 |
0.86 |
| 23 |
B2 |
3231 |
3078 |
18.77 |
43.03 |
0.75 |
0.86 |
| 24 |
B2 |
3157 |
3008 |
11.04 |
108.01 |
0.75 |
0.86 |
| 25 |
B2 |
1313 |
1251 |
33.56 |
0.02 |
0.75 |
0.86 |
| 26 |
B2 |
1169 |
1114 |
6.79 |
0.27 |
0.75 |
0.86 |
| 27 |
B2 |
932 |
888 |
7.07 |
0.07 |
0.75 |
0.86 |
| 28 |
B2 |
903 |
860 |
6.87 |
0.88 |
0.75 |
0.86 |
| 29 |
B2 |
830 |
791 |
6.27 |
0.33 |
0.75 |
0.86 |
| 30 |
B2 |
476 |
453 |
4.26 |
3.36 |
0.75 |
0.86 |
Unscaled Zero Point Vibrational Energy (zpe) 21467.1 cm
-1
Scaled (by 0.9527) Zero Point Vibrational Energy (zpe) 20451.7 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.