Jump to
S1C2
Energy calculated at MP2=FULL/cc-pVTZ
| | hartrees |
|---|
| Energy at 0K | -275.167300 |
| Energy at 298.15K | |
| HF Energy | -274.236019 |
| Nuclear repulsion energy | 117.241165 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at MP2=FULL/cc-pVTZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A1 |
2078 |
1973 |
30.92 |
67.85 |
0.12 |
0.22 |
| 2 |
A1 |
737 |
700 |
10.13 |
11.81 |
0.09 |
0.16 |
| 3 |
A1 |
514 |
488 |
79.15 |
0.47 |
0.16 |
0.28 |
| 4 |
A1 |
80 |
76 |
10.18 |
1.40 |
0.62 |
0.76 |
| 5 |
A2 |
468 |
444 |
0.00 |
2.90 |
0.75 |
0.86 |
| 6 |
B1 |
477 |
453 |
98.55 |
0.14 |
0.75 |
0.86 |
| 7 |
B2 |
2101 |
1994 |
1116.83 |
2.51 |
0.75 |
0.86 |
| 8 |
B2 |
1247 |
1184 |
104.76 |
0.58 |
0.75 |
0.86 |
| 9 |
B2 |
456 |
433 |
9.24 |
2.28 |
0.75 |
0.86 |
Unscaled Zero Point Vibrational Energy (zpe) 4079.4 cm
-1
Scaled (by 0.9494) Zero Point Vibrational Energy (zpe) 3873.0 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
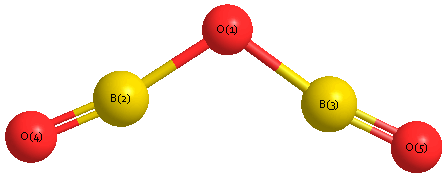
Geometric Data calculated at MP2=FULL/cc-pVTZ
Point Group is C2v
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| O1 |
0.000 |
0.000 |
0.486 |
| B2 |
0.000 |
1.252 |
0.056 |
| B3 |
0.000 |
-1.252 |
0.056 |
| O4 |
0.000 |
2.421 |
-0.278 |
| O5 |
0.000 |
-2.421 |
-0.278 |
Atom - Atom Distances (Å)
| |
O1 |
B2 |
B3 |
O4 |
O5 |
| O1 | | 1.3243 | 1.3243 | 2.5386 | 2.5386 |
B2 | 1.3243 | | 2.5050 | 1.2152 | 3.6884 | B3 | 1.3243 | 2.5050 | | 3.6884 | 1.2152 | O4 | 2.5386 | 1.2152 | 3.6884 | | 4.8414 | O5 | 2.5386 | 3.6884 | 1.2152 | 4.8414 | |
 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| O1 |
B2 |
O4 |
177.028 |
|
O1 |
B3 |
O5 |
177.028 |
| B2 |
O1 |
B3 |
142.092 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Jump to
S1C1
Energy calculated at MP2=FULL/cc-pVTZ
| | hartrees |
|---|
| Energy at 0K | -275.166208 |
| Energy at 298.15K | |
| HF Energy | -274.234896 |
| Nuclear repulsion energy | 116.737217 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at MP2=FULL/cc-pVTZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
Σg |
2086 |
1981 |
0.00 |
87.38 |
0.14 |
0.24 |
| 2 |
Σg |
675 |
641 |
0.00 |
12.42 |
0.13 |
0.23 |
| 3 |
Σu |
2135 |
2027 |
1479.83 |
0.00 |
0.00 |
0.00 |
| 4 |
Σu |
1317 |
1250 |
103.40 |
0.00 |
0.00 |
0.00 |
| 5 |
Πg |
470 |
446 |
0.00 |
3.24 |
0.75 |
0.86 |
| 5 |
Πg |
470 |
446 |
0.00 |
3.24 |
0.75 |
0.86 |
| 6 |
Πu |
446 |
423 |
106.86 |
0.00 |
0.75 |
0.86 |
| 6 |
Πu |
446 |
423 |
106.86 |
0.00 |
0.75 |
0.86 |
| 7 |
Πu |
79i |
75i |
4.12 |
0.00 |
0.00 |
0.00 |
| 7 |
Πu |
79i |
75i |
4.12 |
0.00 |
0.00 |
0.00 |
Unscaled Zero Point Vibrational Energy (zpe) 3943.6 cm
-1
Scaled (by 0.9494) Zero Point Vibrational Energy (zpe) 3744.0 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at MP2=FULL/cc-pVTZ
Point Group is D∞h
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| O1 |
0.000 |
0.000 |
0.000 |
| B2 |
0.000 |
0.000 |
1.310 |
| B3 |
0.000 |
0.000 |
-1.310 |
| O4 |
0.000 |
0.000 |
2.526 |
| O5 |
0.000 |
0.000 |
-2.526 |
Atom - Atom Distances (Å)
| |
O1 |
B2 |
B3 |
O4 |
O5 |
| O1 | | 1.3097 | 1.3097 | 2.5260 | 2.5260 |
B2 | 1.3097 | | 2.6195 | 1.2162 | 3.8357 | B3 | 1.3097 | 2.6195 | | 3.8357 | 1.2162 | O4 | 2.5260 | 1.2162 | 3.8357 | | 5.0519 | O5 | 2.5260 | 3.8357 | 1.2162 | 5.0519 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| O1 |
B2 |
O4 |
180.000 |
|
O1 |
B3 |
O5 |
180.000 |
| B2 |
O1 |
B3 |
180.000 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability