Jump to
S1C2
Energy calculated at MP2=FULL/cc-pVTZ
| | hartrees |
|---|
| Energy at 0K | -357.985431 |
| Energy at 298.15K | -357.990534 |
| HF Energy | -356.686513 |
| Nuclear repulsion energy | 233.431401 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at MP2=FULL/cc-pVTZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3787 |
3595 |
99.30 |
|
|
|
| 2 |
A' |
3779 |
3587 |
95.35 |
|
|
|
| 3 |
A' |
3643 |
3458 |
76.11 |
|
|
|
| 4 |
A' |
1824 |
1732 |
150.73 |
|
|
|
| 5 |
A' |
1806 |
1715 |
371.78 |
|
|
|
| 6 |
A' |
1617 |
1535 |
101.05 |
|
|
|
| 7 |
A' |
1457 |
1384 |
22.27 |
|
|
|
| 8 |
A' |
1338 |
1270 |
68.80 |
|
|
|
| 9 |
A' |
1193 |
1133 |
270.02 |
|
|
|
| 10 |
A' |
1097 |
1041 |
2.85 |
|
|
|
| 11 |
A' |
794 |
754 |
6.77 |
|
|
|
| 12 |
A' |
615 |
584 |
69.05 |
|
|
|
| 13 |
A' |
536 |
509 |
0.17 |
|
|
|
| 14 |
A' |
421 |
400 |
3.96 |
|
|
|
| 15 |
A' |
275 |
261 |
14.34 |
|
|
|
| 16 |
A" |
855 |
812 |
4.35 |
|
|
|
| 17 |
A" |
687 |
652 |
110.67 |
|
|
|
| 18 |
A" |
646 |
613 |
23.23 |
|
|
|
| 19 |
A" |
448 |
425 |
4.72 |
|
|
|
| 20 |
A" |
321 |
305 |
194.72 |
|
|
|
| 21 |
A" |
71 |
67 |
3.25 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 13603.6 cm
-1
Scaled (by 0.9494) Zero Point Vibrational Energy (zpe) 12915.3 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at MP2=FULL/cc-pVTZ
Point Group is Cs
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.000 |
0.745 |
0.000 |
| C2 |
-0.053 |
-0.780 |
0.000 |
| O3 |
-1.094 |
-1.404 |
0.000 |
| O4 |
1.033 |
1.374 |
0.000 |
| O5 |
-1.216 |
1.282 |
0.000 |
| N6 |
1.189 |
-1.297 |
0.000 |
| H7 |
1.308 |
-2.291 |
0.000 |
| H8 |
1.978 |
-0.679 |
0.000 |
| H9 |
-1.080 |
2.241 |
0.000 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
O3 |
O4 |
O5 |
N6 |
H7 |
H8 |
H9 |
| C1 | | 1.5263 | 2.4112 | 1.2096 | 1.3291 | 2.3629 | 3.3058 | 2.4371 | 1.8451 |
C2 | 1.5263 | | 1.2130 | 2.4131 | 2.3676 | 1.3454 | 2.0332 | 2.0335 | 3.1910 | O3 | 2.4112 | 1.2130 | | 3.4988 | 2.6887 | 2.2853 | 2.5599 | 3.1558 | 3.6446 | O4 | 1.2096 | 2.4131 | 3.4988 | | 2.2506 | 2.6759 | 3.6760 | 2.2604 | 2.2840 | O5 | 1.3291 | 2.3676 | 2.6887 | 2.2506 | | 3.5263 | 4.3744 | 3.7476 | 0.9682 | N6 | 2.3629 | 1.3454 | 2.2853 | 2.6759 | 3.5263 | | 1.0013 | 1.0019 | 4.2031 | H7 | 3.3058 | 2.0332 | 2.5599 | 3.6760 | 4.3744 | 1.0013 | | 1.7461 | 5.1226 | H8 | 2.4371 | 2.0335 | 3.1558 | 2.2604 | 3.7476 | 1.0019 | 1.7461 | | 4.2282 | H9 | 1.8451 | 3.1910 | 3.6446 | 2.2840 | 0.9682 | 4.2031 | 5.1226 | 4.2282 | |
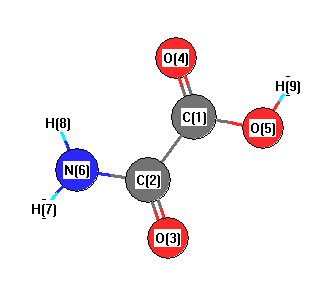 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
O3 |
122.925 |
|
C1 |
C2 |
N6 |
110.581 |
| C1 |
O5 |
H9 |
105.797 |
|
C2 |
C1 |
O4 |
123.352 |
| C2 |
C1 |
O5 |
111.838 |
|
C2 |
N6 |
H7 |
119.364 |
| C2 |
N6 |
H8 |
119.344 |
|
O3 |
C2 |
N6 |
126.494 |
| O4 |
C1 |
O5 |
124.810 |
|
H7 |
N6 |
H8 |
121.291 |
Electronic energy levels
Electronic state
Charges, Dipole, Quadrupole and Polarizability
Jump to
S1C1
Energy calculated at MP2=FULL/cc-pVTZ
| | hartrees |
|---|
| Energy at 0K | -357.992367 |
| Energy at 298.15K | -357.997743 |
| Nuclear repulsion energy | 235.010944 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at MP2=FULL/cc-pVTZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3775 |
3584 |
104.48 |
|
|
|
| 2 |
A' |
3632 |
3448 |
98.46 |
|
|
|
| 3 |
A' |
3607 |
3424 |
152.42 |
|
|
|
| 4 |
A' |
1855 |
1761 |
239.66 |
|
|
|
| 5 |
A' |
1810 |
1718 |
258.22 |
|
|
|
| 6 |
A' |
1620 |
1538 |
59.48 |
|
|
|
| 7 |
A' |
1473 |
1398 |
167.06 |
|
|
|
| 8 |
A' |
1363 |
1294 |
372.34 |
|
|
|
| 9 |
A' |
1233 |
1170 |
4.88 |
|
|
|
| 10 |
A' |
1103 |
1047 |
3.39 |
|
|
|
| 11 |
A' |
820 |
779 |
10.82 |
|
|
|
| 12 |
A' |
638 |
606 |
11.75 |
|
|
|
| 13 |
A' |
553 |
525 |
1.90 |
|
|
|
| 14 |
A' |
412 |
391 |
7.77 |
|
|
|
| 15 |
A' |
277 |
263 |
42.93 |
|
|
|
| 16 |
A" |
854 |
811 |
0.04 |
|
|
|
| 17 |
A" |
782 |
743 |
92.12 |
|
|
|
| 18 |
A" |
678 |
643 |
2.36 |
|
|
|
| 19 |
A" |
480 |
456 |
87.63 |
|
|
|
| 20 |
A" |
386 |
366 |
131.76 |
|
|
|
| 21 |
A" |
121 |
114 |
6.01 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 13734.5 cm
-1
Scaled (by 0.9494) Zero Point Vibrational Energy (zpe) 13039.5 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at MP2=FULL/cc-pVTZ
Point Group is Cs
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.010 |
-0.786 |
0.000 |
| C2 |
0.000 |
0.740 |
0.000 |
| O3 |
-1.068 |
1.336 |
0.000 |
| O4 |
1.018 |
-1.445 |
0.000 |
| O5 |
-1.222 |
-1.273 |
0.000 |
| N6 |
1.222 |
1.272 |
0.000 |
| H7 |
1.343 |
2.266 |
0.000 |
| H8 |
2.010 |
0.650 |
0.000 |
| H9 |
-1.792 |
-0.480 |
0.000 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
O3 |
O4 |
O5 |
N6 |
H7 |
H8 |
H9 |
| C1 | | 1.5264 | 2.3809 | 1.2044 | 1.3246 | 2.3884 | 3.3308 | 2.4627 | 1.8284 |
C2 | 1.5264 | | 1.2236 | 2.4109 | 2.3550 | 1.3328 | 2.0328 | 2.0121 | 2.1680 | O3 | 2.3809 | 1.2236 | | 3.4773 | 2.6141 | 2.2915 | 2.5843 | 3.1541 | 1.9549 | O4 | 1.2044 | 2.4109 | 3.4773 | | 2.2465 | 2.7247 | 3.7257 | 2.3187 | 2.9719 | O5 | 1.3246 | 2.3550 | 2.6141 | 2.2465 | | 3.5284 | 4.3709 | 3.7611 | 0.9776 | N6 | 2.3884 | 1.3328 | 2.2915 | 2.7247 | 3.5284 | | 1.0018 | 1.0034 | 3.4863 | H7 | 3.3308 | 2.0328 | 2.5843 | 3.7257 | 4.3709 | 1.0018 | | 1.7482 | 4.1675 | H8 | 2.4627 | 2.0121 | 3.1541 | 2.3187 | 3.7611 | 1.0034 | 1.7482 | | 3.9669 | H9 | 1.8284 | 2.1680 | 1.9549 | 2.9719 | 0.9776 | 3.4863 | 4.1675 | 3.9669 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
O3 |
119.544 |
|
C1 |
C2 |
N6 |
113.132 |
| C1 |
O5 |
H9 |
104.148 |
|
C2 |
C1 |
O4 |
123.550 |
| C2 |
C1 |
O5 |
111.190 |
|
C2 |
N6 |
H7 |
120.423 |
| C2 |
N6 |
H8 |
118.237 |
|
O3 |
C2 |
N6 |
127.323 |
| O4 |
C1 |
O5 |
125.260 |
|
H7 |
N6 |
H8 |
121.339 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability