Jump to
S1C2
Vibrational Frequencies calculated at MP2=FULL/3-21G*
Geometric Data calculated at MP2=FULL/3-21G*
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Jump to
S1C1
Energy calculated at MP2=FULL/3-21G*
| | hartrees |
|---|
| Energy at 0K | -154.416023 |
| Energy at 298.15K | -154.421423 |
| HF Energy | -154.052464 |
| Nuclear repulsion energy | 104.253425 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at MP2=FULL/3-21G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3264 |
3095 |
6.07 |
|
|
|
| 2 |
A |
3184 |
3019 |
2.34 |
|
|
|
| 3 |
A |
3171 |
3006 |
17.28 |
|
|
|
| 4 |
A |
1663 |
1576 |
2.04 |
|
|
|
| 5 |
A |
1533 |
1453 |
5.27 |
|
|
|
| 6 |
A |
1395 |
1322 |
0.06 |
|
|
|
| 7 |
A |
1117 |
1059 |
0.29 |
|
|
|
| 8 |
A |
1029 |
976 |
3.79 |
|
|
|
| 9 |
A |
959 |
909 |
8.12 |
|
|
|
| 10 |
A |
879 |
834 |
0.34 |
|
|
|
| 11 |
A |
769 |
729 |
5.68 |
|
|
|
| 12 |
A |
285 |
270 |
0.04 |
|
|
|
| 13 |
A |
169 |
161 |
0.01 |
|
|
|
| 14 |
B |
3262 |
3092 |
23.56 |
|
|
|
| 15 |
B |
3177 |
3012 |
7.52 |
|
|
|
| 16 |
B |
3164 |
3000 |
5.54 |
|
|
|
| 17 |
B |
1682 |
1594 |
1.41 |
|
|
|
| 18 |
B |
1518 |
1439 |
0.72 |
|
|
|
| 19 |
B |
1361 |
1291 |
0.39 |
|
|
|
| 20 |
B |
1156 |
1096 |
5.38 |
|
|
|
| 21 |
B |
1047 |
992 |
40.16 |
|
|
|
| 22 |
B |
960 |
910 |
76.89 |
|
|
|
| 23 |
B |
651 |
617 |
7.05 |
|
|
|
| 24 |
B |
472 |
448 |
11.51 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 18933.6 cm
-1
Scaled (by 0.948) Zero Point Vibrational Energy (zpe) 17949.1 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at MP2=FULL/3-21G*
Point Group is C2
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.124 |
0.733 |
0.557 |
| C2 |
-0.124 |
-0.733 |
0.557 |
| C3 |
-0.124 |
1.540 |
-0.492 |
| C4 |
0.124 |
-1.540 |
-0.492 |
| H5 |
0.527 |
1.157 |
1.476 |
| H6 |
-0.527 |
-1.157 |
1.476 |
| H7 |
0.092 |
2.604 |
-0.459 |
| H8 |
-0.566 |
1.158 |
-1.408 |
| H9 |
-0.092 |
-2.604 |
-0.459 |
| H10 |
0.566 |
-1.158 |
-1.408 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
C3 |
C4 |
H5 |
H6 |
H7 |
H8 |
H9 |
H10 |
| C1 | | 1.4869 | 1.3463 | 2.5035 | 1.0894 | 2.2001 | 2.1288 | 2.1255 | 3.4945 | 2.7629 |
C2 | 1.4869 | | 2.5035 | 1.3463 | 2.2001 | 1.0894 | 3.4945 | 2.7629 | 2.1288 | 2.1255 | C3 | 1.3463 | 2.5035 | | 3.0909 | 2.1077 | 3.3628 | 1.0854 | 1.0868 | 4.1444 | 2.9326 | C4 | 2.5035 | 1.3463 | 3.0909 | | 3.3628 | 2.1077 | 4.1444 | 2.9326 | 1.0854 | 1.0868 | H5 | 1.0894 | 2.2001 | 2.1077 | 3.3628 | | 2.5423 | 2.4548 | 3.0842 | 4.2741 | 3.6987 | H6 | 2.2001 | 1.0894 | 3.3628 | 2.1077 | 2.5423 | | 4.2741 | 3.6987 | 2.4548 | 3.0842 | H7 | 2.1288 | 3.4945 | 1.0854 | 4.1444 | 2.4548 | 4.2741 | | 1.8500 | 5.2106 | 3.9088 | H8 | 2.1255 | 2.7629 | 1.0868 | 2.9326 | 3.0842 | 3.6987 | 1.8500 | | 3.9088 | 2.5784 | H9 | 3.4945 | 2.1288 | 4.1444 | 1.0854 | 4.2741 | 2.4548 | 5.2106 | 3.9088 | | 1.8500 | H10 | 2.7629 | 2.1255 | 2.9326 | 1.0868 | 3.6987 | 3.0842 | 3.9088 | 2.5784 | 1.8500 | |
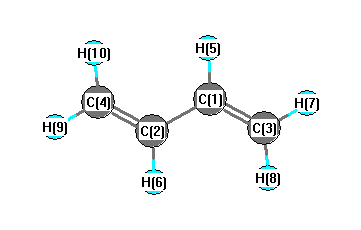 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
C4 |
124.092 |
|
C1 |
C2 |
H6 |
116.441 |
| C1 |
C3 |
H7 |
121.821 |
|
C1 |
C3 |
H8 |
121.388 |
| C2 |
C1 |
C3 |
124.092 |
|
C2 |
C1 |
H5 |
116.441 |
| C2 |
C4 |
H9 |
121.821 |
|
C2 |
C4 |
H10 |
121.388 |
| C3 |
C1 |
H5 |
119.466 |
|
C4 |
C2 |
H6 |
119.466 |
| H7 |
C3 |
H8 |
116.785 |
|
H9 |
C4 |
H10 |
116.785 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability